
Abstract
Background
TP53 mutations in cancer cells often evoke cell invasiveness, whereas fibroblasts show invasiveness in the presence of intact TP53. AMAP1 (also called DDEF1 or ASAP1) is a downstream effector of ARF6 and is essential for the ARF6-driven cell-invasive phenotype. We found that AMAP1 levels are under the control of p53 (TP53 gene product) in epithelial cells but not in fibroblasts, and here addressed that molecular basis of the epithelial-specific function of p53 in suppressing invasiveness via targeting AMAP1.
Methods
Using MDA-MB-231 cells expressing wild-type and p53 mutants, we identified miRNAs in which their expression is controlled by normal-p53. Among them, we identified miRNAs that target AMAP1 mRNA, and analyzed their expression levels and epigenetic statuses in epithelial cells and nonepithelial cells.
Results
We found that normal-p53 suppresses AMAP1 mRNA in cancer cells and normal epithelial cells, and that more than 30 miRNAs are induced by normal-p53. Among them, miR-96 and miR-182 were found to target the 3′-untranslated region of AMAP1 mRNA. Fibroblasts did not express these miRNAs at detectable levels. The ENCODE dataset demonstrated that the promoter region of the miR-183-96-182 cistron is enriched with H3K27 acetylation in epithelial cells, whereas this locus is enriched with H3K27 trimethylation in fibroblasts and other non-epithelial cells. miRNAs, such as miR-423, which are under the control of p53 but not associated with AMAP1 mRNA, demonstrated similar histone modifications at their gene loci in epithelial cells and fibroblasts, and were expressed in these cells.
Conclusion
Histone modifications of certain miRNA loci, such as the miR-183-96-182 cistron, are different between epithelial cells and non-epithelial cells. Such epithelial-specific miRNA regulation appears to provide the molecular basis for the epithelial-specific function of p53 in suppressing ARF6-driven invasiveness.
Similar content being viewed by others


Background
TP53, which encodes the tumor suppressor protein p53, is frequently mutated in human cancers. TP53 mutations (i.e., loss of normal-p53 function) not only promote cell cycle progression, and cell growth and survival, but also evoke invasiveness and mesenchymal phenotypes in various cancer cells [1]. As for the inhibition of invasiveness by p53, the currently prevailing model indicates that p53 induces specific microRNAs (miRNAs) that target mRNAs of transcriptional factors that drive epithelial-mesenchymal transition (EMT-TFs), such as ZEB1, SNAI1, SLUG (SNAI2), and BMI1 [2,3,4]. However, other types of cells, such as bona fide fibroblasts, demonstrate high invasiveness in the presence of intact TP53, and express these EMT-TFs [5]. Thus, some p53-miRNA axes might be specific to epithelial cells, although the molecular bases for such an epithelial-specific function of p53 remains largely elusive [6, 7].
AMAP1 (also called DDEF1 or ASAP1) is a downstream effector of the small GTP-binding protein ARF6 [8]. AMAP1 has multiple protein-protein interaction modules, and can interact with PRKD2 to promote integrin recycling [9], with EPB41L5 to disrupt E-cadherin-mediated cell-cell adhesion [10, 11], and also with cortactin and paxillin to remodel the actin-based cytoskeletal architecture [12]. Thus, AMAP1 is at the core for controlling cell invasiveness under the activity of ARF6, particularly during epithelial-mesenchymal transition (EMT). AMAP1, as well as ARF6, are expressed almost ubiquitously in various types of cells, although their enhanced expression is required to substantially drive cell invasive activity [13,14,15].
The AMAP1 mRNA contains a 5′-terminal oligopyrimidine (TOP)-like sequence at its 5′-untranslated region (UTR), and hence is under the control of mTORC1 (S. Hashimoto et al., submitted). We here show that AMAP1 mRNA is also under the control of p53, in which p53 appears to utilize miRNAs to target the 3’-UTR of this mRNA. Our analysis on the expression of p53-regulatable miRNAs provides insight into the molecular basis by which a specific p53-miRNA axis functions in epithelial cells but not in fibroblasts.
Methods
Cell lines
HEK293T cells, MDA-MB-231 cells, MCF7 cells, and BJ cells were purchased from American Type Culture Collection. MDA-MB-231 cells were cultured in 7.5% CO2 at 37 °C in a 1:1 mixture of Dulbecco’s modified Eagle medium (DMEM) (Invitrogen) and RPMI 1640 (Invitrogen), with 10% fetal calf serum (FCS) (HyClone) and 5% NU serum (BD Biosciences). The p53 derivatives of MDA-MB-231 cells were generated previously [16]. HEK293T cells, MCF7 cells and BJ cells were cultured at 37 °C in DMEM with 10% FCS (GE Healthcare, Illinois, USA). HMLE cells were gifted from Dr. Weinberg (Whitehead Institute, MIT, Cambridge, Massachusetts, USA) and cultured in Mammary Epithelial Cell Growth Medium (MEGM) (Lonza, Maryland, USA). HMLE cells expressing shp53 vectors were generated previously [11].
MiRNA expression profiling
Cells were serum-starved for 16 h, and then left untreated or treated with TGFβ1 (2 ng/mL) for 2 h in the absence of FCS. Total cellular RNAs were then isolated using the QIAGEN RNeasy Mini Kit (QIAGEN, Netherland), according to the manufacturer’s instructions. Microarray analysis of miRNA expression was performed by Toray (Tokyo, Japan) using total cellular RNAs. Color visualization of the data was performed using Java TreeView software.
RNA extraction and quantitative real-time polymerase chain reaction (RT-qPCR)
Total miRNA and mRNA were extracted from cultured cells using the miRNeasy mini kit (QIAGEN). TaqMan gene expression assays (Applied Biosystems:AB, Massachusetts, USA) were used for the analysis of AMAP1 mRNA, and TaqMan miRNA assays (AB) were for the analysis of hsa-miR-96 and hsa-miR182. Glyceraldehyde-3-phosphate dehydrogenase and the U6 small nuclear RNA were used as internal controls. These data were collected by 7300 Real Time PCR System (AB) and the ΔΔCt method was used for relative quantification [17].
Dual luciferase reporter assay
HEK293T cells were transiently transfected using Lipofetamine LTX with pEZX-MT01 target reporter plasmids containing the full length of wild-type AMAP1 3′-UTR and its mutants lacking the miRNA target sequences, all of which were purchased from GeneCopeia (Maryland, USA). These cells were simultaneously transfected with oligonucleotide precursors of hsa-miR-96, hsa-miR-182, or hsa-miR-301a. A total of 5.0 × 104 cells were plated onto a 24-well plate. Luciferase assays were performed 24 h after transfection using Dual-Luciferase Reporter Assay System (Promega, Madison, USA). Firefly luciferase activities were normalized to Renilla luciferase activity for each sample.
Immunoblotting
P53 and β-actin were detected by antibodies purchased from commercial sources (mouse monoclonal anti-TP53, clone #2524, Cell Signaling; mouse monoclonal anti-β-actin, EMD Millipore, Massachusetts, USA). Rabbit polyclonal antibodies against AMAP1 were established as described previously [18]. Peroxidase-conjugated donkey antibodies against mouse or rabbit IgGs were purchased from Jackson ImmunoResearch Laboratories, Inc. All immunoblotting analyses were performed as described previously [16] using ECL Western detection reagents (GE Healthcare, Illinois, USA).
Statistical analysis
All data were statistically analyzed by the unpaired t-test or Spearman rank correlation test using Prism 6.0 software (GraphPad Software, California, USA). A P-value of less than 0.05 was considered to indicate a statistically significant difference between two groups.
Results
TP53 mutations enhance AMAP1 expression in epithelial cells
MDA-MB-231 breast cancer cells express R280K mutant-p53 and have lost the other TP53 allele [19]. We previously generated MDA-MB-231 cells in which endogenous mutant-p53 was silenced (shp53 cells), and shp53 cells expressing normal-p53 (shp53/wt cells) [16]. Western blot analysis of these cells demonstrated that AMAP1 protein levels were largely reduced in the presence of intact-p53 (Fig. 1a). AMAP1 mRNA levels were also significantly suppressed in the presence of intact-p53 (Fig. 1b).

Loss of normal-p53 induces AMAP1 expression in MDA-MB-231 and HMLE cells. a Expression levels of AMAP1 and p53 were assessed by immunoblotting (left). The ratio of AMAP1/Actin was analyzed by densitometry and the results (right) are shown as the mean ± SEM (n = 2). n.s. means no significance. *P < 0.05. b Relative units of AMAP1 mRNA were measured by quantitative-PCR. The mRNA level of MDA-MB-231 cells (parental) was used as a reference value for the other cell lines. The results are shown as the mean ± SEM (n = 3, HMLE series, n = 2, MDA-MB-231). *P < 0.05
To investigate whether the involvement of p53 in the suppression of AMAP1 expression is a general event, we next analyzed non-transformed cells. HMLE cells were generated by the immortalization of primary human normal mammary epithelial cell [20]. These HMLEs expressed the AMAP1 protein at a basal level, similar to that seen in shp53/wt cells (Fig. 1a); and shRNA-mediated silencing of p53 in these cells significantly enhanced AMAP1 expression, both at the protein and mRNA levels (Fig. 1a and b). Therefore, collectively, normal-p53 appeared to function to suppress AMAP1 levels in mammary epithelial cells, regardless of whether they are transformed or not.
P53 induces miR-96 and miR-182 to target AMAP1 mRNA
The expression of various miRNAs is under the control of p53 [21]. To understand the possible mechanisms by which normal-p53 suppresses AMAP1 expression, we then analyzed the expression of miRNAs in MDA-MB-231 cells. In these experiments, we prepared miRNAs from cells cultured at sparse densities, to avoid density-dependent artefacts of miRNA preparation [22, 23]. Thirty-two different miRNAs were found to be expressed at significantly higher levels in shp53/wt cells than in the parental cells and shp53 cells (Fig. 2a). Nine of these miRNAs had nucleotide sequences that were complementary to the 3’-UTR of AMAP1 mRNA (Fig. 2b). Among them, miR-96 and miR-182 levels were negatively correlated with the level of AMAP1 mRNA in the TCGA RNASeq dataset on human primary breast tumors (Fig. 2c and Additional file 1: Figure S1). miR-96 and miR-182 are transcribed as the miR-183-96-182 cistron [24], and it was shown previously that p53 is responsible for the expression of this cistron by its direct binding [25].

miR-96 and miR-182 target AMAP1 mRNA. a Microarray analysis of gene expression in MDA-MB-231 cells (parental) and their p53 derivatives. Green, black, and red indicate transcript levels below, equal to, or above the mean, respectively, as shown at the bottom, in which gene expression intensities are represented in the log 2 scale. b Thirty-two miRNAs were expressed at higher levels in shp53/wt cells than in parental and shp53 cells (a), and 9 of them had homology to the 3’-UTR of the AMAP1 mRNA. c Correlation diagrams between expression levels of miRNA candidates and AMAP1 mRNA are shown. d The interactions among the AMAP1 mRNA 3’-UTR (wt), or its mutant (mt) and miR-96, miR-182, or miR-301a, were assessed by luciferase reporter assay in HEK293T cells. Luciferase activities were measured and normalized to Renilla luciferase activity. The results are shown as the mean ± SEM (n = 3). *P < 0.05, **P < 0.01. e mRNA expression levels of two miRNA candidates in MDA-MB-231 cells and their p53 derivatives, and HMLE cells and their p53-knockdown cells were measured by quantitative RT-PCR. The results are shown as the mean ± SEM (n = 2). n.s. means no significance. *P < 0.05
We then generated a reporter gene, in which the firefly luciferase gene is fused to the AMAP1 3’-UTR, and found that miR-96 and miR-182 have the ability to target this 3’-UTR in a reconstituted system using HEK293T cells (Fig. 2d). As a control, we analyzed miR-301a. miR-301a was induced by normal-p53 and had a sequence complementary to the AMAP1 3’-UTR, whereas the TCGA database did not support a reciprocal association between this miRNA and AMAP1 mRNA (Fig. 2a-c). We found that miR-301a is ineffective in targeting the AMAP1 3’-UTR in the reconstitution system (Fig. 2d). The suppression of miR-96 and miR-182 expression upon the loss of intact-p53 was also observed in HMLE cells (Fig. 2e), and we also confirmed statistically significant changes in miR-96 and miR-182 expression levels in MDA-MB-231 cells, depending on the p53 status (Fig. 2e). Therefore, miR-96 and miR-182 are likely to be involved in the suppression of AMAP1 mRNA levels in response to normal-p53.
The p53-miR-96/182 axis is specific to epithelial cells and does not exist in fibroblasts
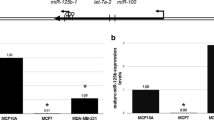
MCF7 breast cancer cells express normal-p53 and are weakly invasive [26]. Consistently, MCF7 cells expressed the AMAP1 protein, as well as AMAP1 mRNA at much lower levels than MDA-MB-231 cells (Fig. 3a and b). On the other hand, normal human fibroblasts bearing intact TP53, such as BJ cells, expressed AMAP1 mRNA and protein at higher levels than MCF7 cells (Fig. 3a and b). We found that BJ cells do not notably express miR-96 and miR-182 (Fig. 3a).

Expression and Epigenomic status of miR-96, miR-182, and AMAP1. a Expression levels of miR-96, miR-182, and AMAP1 in MDA-MB-231 cells, MCF7 cells, and BJ cells were measured by quantitative RT-PCR. The unpaired t-test was performed (miR-96: n = 2; miR-182, AMAP1: n = 3) and the results are shown as the mean ± SEM. **P < 0.01. #: The mRNA level of BJ cell was under the detection limit. b Expression levels of AMAP1 were assessed by immunoblotting. c The ENCODE data of miR-183-96-182 and miR-423 loci by UCSC Genome Browser are shown. Definitions of the colors are given at the top. hESC: human embryonic stem cells, HMEC: human mammary epithelial cells, NHEK: normal human epidermal keratinocytes, NHLF: normal human lung fibroblasts, GM12878: B-lymphocyte transformed by Epstein-Barr virus, K562: highly undifferentiated and granurocytic myelogenous leukemia cells, HepG2:hephepatocytellular carcinoma cells, HUVEC: human umbilical vein endothelial cells, HSMM: human skeletal muscle myoblasts
A dataset of the ENCODE project [27, 28] then indicated that histone modification around the promoter region of the miR-183-96-182 cistron is enriched with H3K27 trimethylation (H3K27me3) in fibroblasts, as well as in other non-epithelial cells, whereas this locus is enriched with H3K27 acetylation (H3K27ac) in epithelial cells (Fig. 3c). Thus, collectively, transcription of this cistron appeared to be epigenetically suppressed in fibroblasts, although it can be transcribed in epithelial cells.
Not all p53-miRNA axes are specific to epithelial cells
The above results suggested an epithelial specificity of the p53-miR-96/182 axis. Normal p53 is known to induce many different types of miRNAs, as we have also shown above (see Fig. 2a). We were finally interested in understanding whether the induction of miRNAs by p53 is specific to epithelial cells, and does not occur in fibroblasts. miR-423 is highly induced in the presence of p53 in MDA-MB-231 cells, but does not have nucleotide sequences complementary to the AMAP1 mRNA 3’-UTR (see Fig. 2a and b). We found that histone modification of this locus was similar both in epithelial cells and fibroblasts, and was categorized as an active promoter (Fig. 3c). Likewise, histone modifications of the gene loci of some other miRNAs, which are under the control of normal-p53 in MDA-MB-231 cells, were not necessarily suppressed in fibroblasts (Additional file 2: Figure S2). Therefore, we concluded that not all p53-miRNA axes are specific only to epithelial cells.
Conclusions
In this study, we showed that p53 acts to restrict AMAP1 mRNA levels in epithelial cells, but not in fibroblasts. AMAP1 mRNA contains a 5’-TOP-like sequence, and is hence under the control of mTORC1, as mentioned earlier. Therefore, a double safeguard system appears to exist to prevent high expression levels of AMAP1 protein, to prevent the cell-invasive phenotype from readily appearing, by targeting the 5’-UTR and 3’-UTR of AMAP1 mRNA. Thus, normal-p53 appears to be an epithelial-specific safeguard preventing cell invasiveness, under conditions where mTORC1 becomes activated such as during active cell proliferation.
MiR-96 and miR-182 are first transcribed as a miR-183-96-182 cistron, as mentioned earlier. The basic functions of miRNAs include coordination of the expression of various mRNAs, to orchestrate protein levels as required for specific cellular functions. In this regard, it is interesting to note that miR-96 also targets Foxf2 and Ezrin mRNAs, to block the invasion and metastasis of lung cancers and renal cancers [29, 30]. miR-183 was also reported to be involved in inhibition of the invasion and metastasis of different cancers, such as lung, breast, and osteosarcoma [31,32,33]. On the other hand, normal-p53 may induce miR-200c to suppress the invasiveness of HMECs and MCF12A cells, in which miR-200c targets ZEB1 to block EMT [2]. We found that miR-200c is not complementary to AMAP1 mRNA and not notably induced by p53 in MDA-MB-231 cells (data not shown). Therefore, p53 appears to have various routes to suppress invasiveness, via different miRNAs in different epithelial cells. On the other hand, we do not know why cancer cells, such as MDA-MB-231 cells, have very low but detectable levels of miR-96/182 expression even in the absence of intact p53 (i.e., leaky expression of miR-96/182 in the parental cells and shp53 cells), whereas normal epithelial cells have undetectable levels of miR-96/182 upon the loss of intact p53.
Our results indicated that the induction of miR-96 and miR-182 by p53 is an event specific to epithelial cells. Such epithelial cell-specific miRNA induction by p53 appeared to be predetermined by the epigenetic regulation of these miRNA gene loci. Moreover, not all miRNAs that are under the control of p53 in epithelial cells are specific to epithelial cells. Thus, our results give rise to the following questions: 1) when and what determine the epigenetic regulation of certain miRNAs, such as the miR-183-96-182 cistron, to confer p53 the ability to block invasiveness in epithelial cells; and 2) whether or not epithelial cells increase AMAP1 mRNA levels in the presence of intact-p53, to gain high invasiveness, such as during EMT, in addition to the mTORC1-mediated translational upregulation of this mRNA.
Abbreviations
- EMT:
-
Epithelial-mesenchymal transition
- ENCODE:
-
Encyclopedia of DNA elements
- miRNA:
-
Micro ribonucleic acid
- mTORC1:
-
Mechanistic target of rapamycin complex 1
- RT-qPCR:
-
Real time quantitative polymerase chain reaction
- TCGA:
-
The cancer genome atlas
- TOP:
-
Terminal oligopyrimidine
- UTR:
-
Untranslated region
References
Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–78.
Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–23.
Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208:875–83.
Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21–45.
Baulida J. Epithelial-to-mesenchymal transition transcription factors in cancer-associated fibroblasts. Mol Oncol. 2017;11:847–59.
Oikawa T, Otsuka Y, Sabe H. p53-dependent and -independent epithelial integrity: beyond miRNAs and metabolic fluctuations. Cancers (Basel). 2018;10.
Oikawa T, Otsuka Y, Onodera Y, Horikawa M, Handa H, Hashimoto S, Suzuki Y, Sabe H. Necessity of p53-binding to the CDH1 locus for its expression defines two epithelial cell types differing in their integrity. Sci Rep. 2018;8:1595.
Onodera Y, Nam JM, Sabe H. Intracellular trafficking of integrins in cancer cells. Pharmacol Ther. 2013;140:1–9.
Onodera Y, Nam JM, Hashimoto A, Norman JC, Shirato H, Hashimoto S, Sabe H. Rab5c promotes AMAP1-PRKD2 complex formation to enhance beta1 integrin recycling in EGF-induced cancer invasion. J Cell Biol. 2012;197:983–96.
Hirano M, Hashimoto S, Yonemura S, Sabe H, Aizawa S. EPB41L5 functions to post-transcriptionally regulate cadherin and integrin during epithelial-mesenchymal transition. J Cell Biol. 2008;182:1217–30.
Hashimoto A, Hashimoto S, Sugino H, Yoshikawa A, Onodera Y, Handa H, Oikawa T, Sabe H. ZEB1 induces EPB41L5 in the cancer mesenchymal program that drives ARF6-based invasion, metastasis and drug resistance. Oncogene. 2016;5:e259.
Hashimoto S, Hirose M, Hashimoto A, Morishige M, Yamada A, Hosaka H, Akagi K, Ogawa E, Oneyama C, Agatsuma T, et al. Targeting AMAP1 and cortactin binding bearing an atypical src homology 3/proline interface for prevention of breast cancer invasion and metastasis. Proc Natl Acad Sci U S A. 2006;103:7036–41.
Menju T, Hashimoto S, Hashimoto A, Otsuka Y, Handa H, Ogawa E, Toda Y, Wada H, Date H, Sabe H. Engagement of overexpressed Her2 with GEP100 induces autonomous invasive activities and provides a biomarker for metastases of lung adenocarcinoma. PLoS One. 2011;6:e25301.
Sato H, Hatanaka KC, Hatanaka Y, Hatakeyama H, Hashimoto A, Matsuno Y, Fukuda S, Sabe H. High level expression of AMAP1 protein correlates with poor prognosis and survival after surgery of head and neck squamous cell carcinoma patients. Cell Communication and Signaling. 2014;12:17.
Hashimoto S, Mikami S, Sugino H, Yoshikawa A, Hashimoto A, Onodera Y, Furukawa S, Handa H, Oikawa T, Okada Y, et al. Lysophosphatidic acid activates Arf6 to promote the mesenchymal malignancy of renal cancer. Nat Commun. 2016;7:10656.
Hashimoto A, Oikawa T, Hashimoto S, Sugino H, Yoshikawa A, Otsuka Y, Handa H, Onodera Y, Nam JM, Oneyama C, et al. P53- and mevalonate pathway-driven malignancies require Arf6 for metastasis and drug resistance. J Cell Biol. 2016;213:81–95.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–8.
Onodera Y, Hashimoto S, Hashimoto A, Morishige M, Mazaki Y, Yamada A, Ogawa E, Adachi M, Sakurai T, Manabe T, et al. Expression of AMAP1, an ArfGAP, provides novel targets to inhibit breast cancer invasive activities. EMBO J. 2005;24:963–73.
Wasielewski M, Elstrodt F, Klijn JG, Berns EM, Schutte M. Thirteen new p53 gene mutants identified among 41 human breast cancer cell lines. Breast Cancer Res Treat. 2006;99:97–101.
Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65.
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4.
Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108:11930–5.
Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, Gregory RI. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell. 2014;156:893–906.
Ma Y, Liang AJ, Fan YP, Huang YR, Zhao XM, Sun Y, Chen XF. Dysregulation and functional roles of miR-183-96-182 cluster in cancer cell proliferation, invasion and metastasis. Oncotarget. 2016;7:42805–25.
Kouri FM, Hurley LA, Daniel WL, Day ES, Hua Y, Hao L, Peng C-Y, Merkel TJ, Queisser MA, Ritner C, et al. miR-182 integrates apoptosis, growth. and differentiation programs in glioblastoma Genes & Development. 2015;29:732–45.
Wang S-P, Wang W-L, Chang Y-L, Wu C-T, Chao Y-C, Kao S-H, Yuan A, Lin C-W, Yang S-C, Chan W-K, et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol. 2009;11:694.
Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Systematic analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9.
The EPC. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57.
Kundu ST, Byers LA, Peng DH, Roybal JD, Diao L, Wang J, Tong P, Creighton CJ, Gibbons DL. The miR-200 family and the miR-183~96~182 cluster target Foxf2 to inhibit invasion and metastasis in lung cancers. Oncogene. 2016;35:173–86.
Yu N, Fu S, Liu Y, Xu Z, Liu Y, Hao J, Wang B, Zhang A. miR-96 suppresses renal cell carcinoma invasion via downregulation of Ezrin expression. J Exp Clin Cancer Res. 2015;34:107.
Wang G, Mao W, Zheng S. MicroRNA-183 regulates Ezrin expression in lung cancer cells. FEBS Lett. 2008;582:3663–8.
Lowery AJ, Miller N, Dwyer RM, Kerin MJ. Dysregulated miR-183 inhibits migration in breast cancer cells. BMC Cancer. 2010;10:502.
Zhu J, Feng Y, Ke Z, Yang Z, Zhou J, Huang X, Wang L. Down-regulation of miR-183 promotes migration and invasion of osteosarcoma by targeting Ezrin. Am J Pathol. 2012;180:2440–51.
Acknowledgements
We thank Y. Kado for her technical assistance, A. Oda for her secretarial support, and H.A. Popiel for her critical reading of the manuscript.
Funding
This study was supported in part by JSPS KAKENHI Grant Numbers 18H02608 and 23112008 to H. Sabe.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
HS designed the study. HH, AH, SH, HSu, and TO performed the experiments and HH, AH, SH, HSu and HS analyzed the data. HH, AH, and HS wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Handa, H., Hashimoto, A., Hashimoto, S. et al. Epithelial-specific histone modification of the miR-96/182 locus targeting AMAP1 mRNA predisposes p53 to suppress cell invasion in epithelial cells. Cell Commun Signal 16, 94 (2018). https://doi.org/10.1186/s12964-018-0302-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-018-0302-6