
Abstract
Infections with the human pathogen Helicobacter pylori (H. pylori) are closely associated with the development of inflammatory disorders and neoplastic transformation of the gastric epithelium. Drastic changes in the micromilieu involve a complex network of H. pylori-regulated signal transduction pathways leading to the release of proinflammatory cytokines, gut hormones and a wide range of signaling molecules. Besides controlling embryonic development, the Hedgehog/GLI signaling pathway also plays important roles in epithelial proliferation, differentiation, and regeneration of the gastric physiology, but also in the induction and progression of inflammation and neoplastic transformation in H. pylori infections. Here, we summarize recent findings of H. pylori-associated Hedgehog/GLI signaling in gastric homeostasis, malignant development and the modulation of the gastric tumor microenvironment.
Similar content being viewed by others

Background
Although the incidence of gastric cancer steadily declined in the last 20 years, stomach cancer is still the second leading cause for cancer-related deaths worldwide [1]. As the major causative agent for gastric cancer, the human bacterial pathogen Helicobacter pylori (H. pylori) has been identified, which is responsible for more than 70% of gastric adenocarcinomas (non-cardia gastric cancers) and also for other gastric disorders including chronic gastritis, ulceration of the stomach and duodenum, and lymphomas of the mucosa-associated lymphoid tissue (MALT) system [2, 3]. According to the strong association between infections with H. pylori and neoplastic transformations in the human stomach, H. pylori has been classified as a class-I carcinogen, representing the strongest known risk factor for gastric cancer [4].
Gastric cancer can be histologically differentiated between diffuse or intestinal types and both are linked to chronic H. pylori infections in humans. The pathogenesis of the diffuse-type carcinoma is less well understood, but has been frequently associated with the loss of expression of the cell adhesion molecule and tumor suppressor E-cadherin (CDH1). Loss of E-cadherin function is often the consequence of cdh1 germline mutations and could also be linked to sporadic mutations or promoter hypermethylation. Tumor cells exhibiting CDH1 malfunction and subsequently loss of intercellular adhesions tend to invade adjacent tissues and are considered as more aggressive compared to tumor cells of the intestinal type [5, 6]. Gastric cancer of the intestinal type typically involves a series of sequential processes, which are strongly linked to H. pylori infections. According to the Correa’s cascade, chronic active inflammation in response to persistent H. pylori infection represents the initial phase in carcinogenesis followed by chronic atrophic gastritis, intestinal metaplasia, dysplasia, and finally invasive carcinoma [6]. During gastric carcinogenesis, genetic abnormalities accumulate and may involve mutations in the APC, TP53, and KRAS genes, but also hypermethylation and microsatellites were detected [7, 8].
Normally, H. pylori infection is acquired in childhood, and persists for the patient’s lifetime if not treated with antibiotics. Although infections with H. pylori are prevalent, only approximately 1–3% of the patients develop gastric cancer [3]. The clinical outcome strongly depends on the crosstalk between strain-specific bacterial virulence factors, genetic predispositions of the host, alterations of the stem cell niche, microbiota and environmental influences. In this context, implications of gene polymorphisms have been described including interleukins and antagonistic receptors such as IL1B, IL10, IL1RN and TNF-alpha [9, 10]. Environmental factors include smoking, high salt consumption, processed meat or alcohol as possible risk factors for gastric cancer (Fig. 1a). In contrast, consumption of fresh fruits and vegetables has been associated with reduced cancer risk. The major bacterial determinant in the risk of developing gastric cancer is represented by the cytotoxin-associated gene pathogenicity island (cagPAI). The cagPAI is a 40 kB DNA insertion element consisting of 27-31 genes that encode proteins important for the structure and function of a highly-specialized type IV secretion system (T4SS) [11]. The T4SS translocates the only known effector protein cytotoxin-associated gene A (CagA) into the cytoplasm of infected gastric epithelial and immune host cells where it is tyrosine phosphorylated by non-receptor tyrosine kinases of the Src and Abl kinase families [12–14] and derails cancer-associated signal transduction pathways [15, 16]. In fact, infections with CagA-positive H. pylori strains have been strongly correlated with the development of severe inflammatory responses and subsequently gastric cancer [17]. It has been suggested that in comparison to cagA-negative isolates, H. pylori strains expressing CagA increase the risk of distal gastric adenocarcinoma twofold [18]. Using in vivo animal models, CagA translocation has been suggested to play an important role in the induction of gastric cancer [19, 20]. Transgenic mice systemically expressing CagA underlined this observation through the finding that CagA increased gastric epithelial cell proliferation rates and carcinomas [21]. Besides CagA, the expression of additional factors was described as further important bacterial determinant in the development of gastric cancer, such as vacuolating cytotoxin A (VacA) [22, 23], adhesion factors as blood group antigen-binding adhesin (BabA) [24] and sialic acid-binding adhesin (SabA) [25].

Model of the gastric physiology and cancer development. a Infection with H. pylori has been strongly associated with the development of the diffuse type and the intestinal type of gastric cancer. The diffuse type is often accompanied by the loss of E-cadherin (CDH1) expression. The development of the intestinal type of gastric cancer is associated with chronic gastritis, atrophy, and intestinal metaplasia as precursors of dysplastic changes. Mutations, hypermethylation, and microsatellites, but also environmental factors are implicated in the carcinogenic process. In this model, chronic active inflammation represents the initial phase in carcinogenesis via alterations of epithelial apoptosis, cell proliferation, recruitment of BM-MSCs, dedifferentiation processes and induced invasive growth of neoplastic cells. b The gastric physiology is established by the coordinated action of paracrine factors and hormones. The epithelium contains parietal cells, D cells, G cells and circulating enterochromaffin-like (ECL) cells. The release of gastric acid by parietal cells is stimulated by ECL-secreted histamine and gastrin expressed by G cells. D cells produce the negative regulator somatostatin, which blocks acid secretion via direct effects on parietal cells and through the inhibition of histamine and gastrin release
H. pylori induced gastric cancer and the tumor microenvironment
The mechanism of how H. pylori can induce gastric cancer is not well understood. H. pylori induces a plethora of different signal transduction processes that trigger a complex multi-step process leading to inflammation and carcinogenesis [26–29]. Normally, these pathways critically control cellular responses such as proliferation, apoptosis, epithelial dedifferentiation and motility, thereby regulating tissue homeostasis (Fig. 1a). So far, most studies of H. pylori induced cancer have focused on specific cell types, although the interplay between different cell types ranging from gastric epithelial cells, glands, immune cells, to stem cells is crucially important for the development and progression of H. pylori-associated carcinogenesis [30–32].
H. pylori associated gastric cancer is characterized by a chronic inflammatory phenotype, where the contribution and interaction of bacterial virulence factors and the host immune system account for oncogenic transformation (for review see [30] and references therein). This becomes evident at the molecular as well as cellular level. For instance, H. pylori has been reported to activate the key inflammatory regulator nuclear factor kappa B (NF-κB), resulting in the activation and enhancement of cytokine signaling including IL-8 and TNF-alpha [33–38]. Further, IL11 mediated activation of STAT3, an important regulator of inflammation and driver of carcinogenesis, is a hallmark of about 50 percent of gastric cancers and has been shown to contribute to tumor growth within an inflammatory setting [39, 40]. At the cellular level, myeloid and lymphocytic cells frequently infiltrate malignant lesions. Tumor-associated macrophages (TAM) promote malignant progression and the degree of TAM-infiltration induced by a variety of chemoattractant factors correlates with tumor progression and clinical disease stage [41–43]. Also, the number of immunosuppressive regulatory T-cells (Tregs) is enhanced in tumor-draining lymph nodes and peripheral blood of gastric cancer patients and the number of Tregs inversely correlates with the survival of patients [44–49]. Besides cells of the innate and adaptive immune system, the tumor microenvironment is to a large degree made up of cancer-associated fibroblasts (CAF) that develop in response to the interplay of cancer cells with their stromal environment. CAF support cancer growth and progression by producing pro-tumorigenic and -metastatic factors including pro-angiogenic signals [50–53]. Thus, a detailed understanding of oncogenic signaling pathways within the tumor and stromal compartments, particularly also in inflammatory and immunosuppressive cell types is needed to guide the design of novel combination therapies that may involve strategies blocking both immunosuppressive and pro-tumorigenic inflammatory signals in the tumor microenvironment together with targeted inhibition of oncogenic driver cues in gastric cancer cells.
Gastric physiology and Hedgehog/GLI signaling in gastric cancer
Dependent on the region in the human stomach, the gastric epithelial lining forms foveolae consisting of different types of cells and glands, including mucous, endocrine, and undifferentiated cells (Fig. 1b), which coordinate the complex gastric physiology by a balanced micromilieu. Embedded within undifferentiated epithelial cells, D cells, G cells and circulating enterochromaffin-like (ECL) cells release regulatory molecules controlling the production of gastric acid by parietal cells [54, 55]. Histamine is released from ECL cells, the hormone gastrin is released by G cells, and the hormone somatostatin is secreted by D cells. In a paracrine manner, histamine stimulates parietal cells to produce gastric acid. Gastrin is involved in acid secretion, stimulating histamine release from ECL cells. As a negative regulator, somatostatin release is stimulated when the pH in the stomach is too low. Then it blocks acid secretion via direct effects on parietal cells, but also through the inhibition of histamine and gastrin release [54, 55] (Fig. 1b). This sensitive balance of intercellular communication can be crucially interrupted by infections with H. pylori through manifold mechanisms [56, 57]. As an additional important part of changes in the gastric tumor microenvironment, H. pylori stimulates a wide range of proinflammatory mediators employing a highly complex network of a wide range of diverse signaling pathways [16, 58, 59]. In fact, relatively little is known about the detailed molecular processes and signals operating during the early and later stages of gastric cancer in response to H. pylori infection and chronic inflammation. In recent years, several oncogenic pathways including the wingless-type MMTV integration site family (Wnt)/beta catenin, NF-κB and Hedgehog/GLI (HH/GLI) signaling pathway have been implemented in the complex network of diverse molecular mechanisms leading to gastric cancer [60]. The implication of HH/GLI signaling in gastric cancer has, therefore, opened the possibility of HH/GLI targeting as a novel therapeutic approach.
The HH/GLI pathway, first discovered in a mutagenesis screen for embryonic patterning mutants of the fruit fly [61], is a crucial developmental regulatory signal that has been highly conserved throughout various phyla. During the past years, HH/GLI signaling has attracted substantial interest by tumor biologist and oncologist because of its widespread hyperactivation and oncogenic activity in a variety of human malignancies. In fact, HH/GLI signaling and its target genes control the major hallmarks of cancer and cancer stem cells including proliferation, survival, metastasis, angiogenesis and self-renewal, making this signaling pathway a promising target for therapies [62–66].
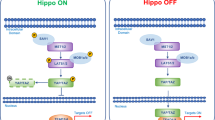
The HH/GLI pathway is a highly complex signal transduction process involving numerous regulatory factors and control mechanisms located in different cellular compartments. In a nutshell, during the off-state canonical HH/GLI signaling is actively repressed via the unliganded, twelve-pass transmembrane HH receptor patched (PTCH) (Fig. 2a). PTCH prohibits the pathway activator Smoothened (SMO), a G-protein coupled receptor-like protein, from entering the primary cilium. In this repressed state, the negative pathway regulator suppressor of fused (SUFU) sequesters the first-line effector proteins, glioblastoma-associated-protein 2 and 3 (GLI2, GLI3) in the cytoplasm at the base of the primary cilium. The formation of the SUFU-GLI protein complex allows the sequential phosphorylation of the GLI proteins by protein kinase A (PKA), glycogen synthase kinase 3 beta (GSK3β) and casein kinase 1 (CK1) [67, 68]. Upon phosphorylation, GLI2 and GLI3 are ubiquitinylated and partially degraded by the proteasome located at the base of the primary cilium [69]. While partial degradation of GLI2 is rather inefficient, GLI3 is known to abundantly form stable transcriptional repressors upon proteolytic removal of the C-terminal portion harboring the transactivation domain [70, 71]. Thus, proteolytic processing yields a GLI repressor form (GLIR) that prevents and shuts off HH target gene expression (for more extensive reviews on HH/GLI signaling see [72–78]).

A simplified schematic depiction of the canonical hedgehog (HH) signal transduction pathway within the primary cilium. a During the absence of the HH ligand the pathway is continuously repressed by PTCH and GPR161 located at the base of the primary cilium. In its unliganded state the HH receptor PTCH prevents SMO, the crucial GLI activator, from entering the plasma membrane of the primary cilium and thereby from executing its effector function (1). Furthermore, GPR161 increases cAMP levels, promoting the phosphorylation of the GLI transcription factors, which are sequestered in a repressive complex with SUFU and IFT proteins at the base of the primary cilium, by PKA and subsequently by GSK3β and CK1 (2). This phosphorylation leads to partial proteasomal degradation of GLI2 and GLI3 and repressor formation, predominantly GLI3R (3). Thereupon, GLI3R enters the nucleus and represses target gene transcription (4). b The binding of the HH ligand to its receptor PTCH is promoted by GAS1, BOC and CDO, whereas HHIP competes with PTCH for ligand binding (5). When HH binds to PTCH the repression of the pathway is relieved by internalization and lysosomal degradation of the receptor-ligand-complex (6). This allows SMO to enter the primary cilium (7) and to be activated by cholesterol, which triggers a conformational change (indicated by the schematic cholesterol structure in black and the asterisk (SMO*)). GPR161 whereas is removed from the plasma membrane (8). When SMO* and the SUFU-GLI complex co-localize at the tip of the primary cilium, upon the directed transport via IFT proteins along the microtubules, the GLI transcription factors are activated by SMO* and dissociate from the complex (9). The full-length activator forms of GLI2 and GLI3 enter the nucleus and drive target gene transcription (10)
The canonical HH/GLI pathway is activated via binding of the HH ligand to the receptor PTCH (Fig. 2b). Ligand binding abrogates the repressive action of PTCH, leading to the internalization of the receptor-ligand complex and its subsequent degradation in lysosomes. HH ligand binding is influenced by the presence of distinct co-receptors: growth arrest specific 1 (GAS1), cell adhesion molecule-related/down-regulated by oncogenes (CDO) and brother of CDO (BOC) support the binding of HH to PTCH, while hedgehog interacting protein (HHIP) competes with PTCH for the HH ligand [79–82]. The removal of PTCH triggers the entry of SMO into and the exit of GPR161 from the primary cilium. SMO is either shifted laterally within the plasma membrane or enters the cilium from intracellular vesicles [83]. There is evidence that the removal of GPR161 is sufficient to prevent GLIR formation, most likely because of a reduced PKA activity [84–86]. GLI activation, however, is crucially dependent on the activation and the correct localization of SMO. Although the specific signal, which activates and represses SMO in response to HH, has not yet been identified, recent work by the Rohatgi group has shed light on the regulatory role of conformational changes of SMO for signal transduction. These studies revealed that cholesterol binding to the extracellular SMO domains stabilizes a conformation that promotes responsiveness towards activating stimuli [87, 88].
When the pathway is activated the GLI-SUFU complex is transported from the base to the tip of the primary cilium. It has been shown that only upon the co-localization of active SMO and GLIs at the tip of the primary cilium, full-length GLI2 and GLI3 are released from SUFU. The full-length GLIs then translocate into the nucleus to activate target gene transcription [69, 89, 90]. Upon GLI activation, positive as well as negative feedback loops are elicited to balance the strength and the duration of pathway activation. GLI1 encodes a second-line but critical pathway amplifier that is directly induced by GLI2 [91, 92]. GLI1 potently amplifies HH/GLI signaling by activating and/or enhancing the expression of a battery of HH target genes. Fine-tuning of the response to GLI activity further depends on interactions with co-factors, post-translational modifications including phosphorylation and acetylation as well as on the differential stability and degradation of the GLIs [93–98]. In addition, different target genes display different sensitivities towards GLI activator (GLIA) and GLIR levels as a consequence of GLI binding site variations with distinct affinities, adding another regulatory layer for the precise determination of the response to the so-called GLI-code [99–102].
Further, there is a steadily increasing list of mechanisms accounting for SMO-independent regulation of GLI activity and expression. Of note, these non-canonical HH/GLI signals have been repeatedly reported in cancer cells, integrating the HH/GLI pathway in the complex web of oncogenic signals but also accounting for resistance to clinical inhibitors targeting SMO, which has become a major challenge for the use of Hedgehog pathway inhibitors in oncology [103–110] (for comprehensive reviews see [102, 111, 112]).
HH/GLI targeting as therapeutic option in gastric cancer – challenges and considerations
Despite substantial efforts of biotech and pharmaceutical companies to develop efficient HH pathway inhibitors, the clinical success of anti-HH therapies has mainly been limited to non-melanoma skin and brain cancers, while other clinical trials using HH/GLI inhibitors for the treatment of solid cancers with high medical need yielded largely disappointing results [113–118]. Although these failed trials were based on sound preclinical evidence supporting a key role of HH/GLI signaling in malignant progression of various cancer entities [119–123], the unforeseeable complexity of HH/GLI signal regulation within the tumor and its microenvironment as well as the frequent development of a priori and/or acquired drug resistance have recently challenged the concept of HH/GLI targeting in oncology [124, 125]. We outline two examples – HH/GLI signaling in pancreatic and colorectal cancer - to emphasize the strict need for a very careful and comprehensive analysis of the oncogenicity of the HH/GLI pathway within the complex interplay of cancer cells with their microenvironment and the immune system, in order to develop multi-modal therapeutic protocols that may be successful in the future treatment of gastric cancer.
First evidence based on in vitro and xenograft models suggested a crucial tumor-cell autonomous role of canonical HH/GLI signaling in pancreatic cancer [119, 126]. However, this concept has recently been challenged by findings showing in vivo activation of HH/GLI signaling in the stromal rather than tumor cell compartment. Strikingly, inhibition of HH/GLI signaling in the tumor stroma of pancreatic cancer led to enhanced tumor growth rather than a therapeutic effect, reflecting the discouraging outcome of anti-HH trials in pancreatic cancer patients [127–129]. By contrast, non-canonical activation of the GLI transcription factors mediating HH/GLI signaling in the nucleus of pancreatic cancer cells is essential for tumor initiation and disease progression [130, 131]. This suggested that direct targeting of oncogenic GLI proteins - while also maintaining the protective effect of the stromal compartment - may prove a successful therapeutic strategy within a multi-modal combination treatment protocol.
Similarly, the initial enthusiasm about HH targeting for the treatment of colorectal cancer faded rapidly, when clinical trials with HH antagonists did not show any significant therapeutic benefit. This may to some extent be due to the fact that most preclinical models used for studying the oncogenic effect of HH/GLI signaling did not take into account the cellular and molecular complexity of the tumor microenvironment and the vivid cross-talk between tumor cells, the tumor stroma and the immune system. Like in pancreatic cancer, it has recently been shown that canonical HH/GLI signaling in colon cancer is strongly activated in the stromal rather than the tumor cell compartment providing a cancer-protective activity. Intriguingly, HH/GLI signaling in the stromal compartment of mouse colon cancer models reduces tumor development by modifying BMP signaling in colon cancer cells and by dampening inflammatory signaling in colitis-associated cancer models [132, 133]. Given the distinct functions of HH/GLI signaling within the heterogeneous cellular context of the tumor and its microenvironment, a precise understanding of HH/GLI signaling in the context of gastric cancer is mandatory for the future evaluation of the therapeutic potential of HH/GLI targeting.
Hedgehog signaling as a crucial mediator in gastric physiology and disease
The role of the HH/GLI signaling pathway in gastric homeostasis has been established in several recent studies (for reviews see [134–136]). The expression of the hedgehog family member sonic hedgehog (SHH) is required to shape the mucosal layer but has to be tightly controlled during the development of the gastric glandular epithelium [135, 137]. Furthermore, SHH expression appears to be crucial for gastric tissue repair [138] and for the maintenance of the functional morphology and the regulation of secretory functions of gastric glands in adult mice [134]. There is evidence that SHH production and reception by parietal cells is required to maintain the acid and gastrin secretion in the stomach at physiological levels. Furthermore, the SHH concentration gradient established by the parietal cells, located in the central region of the gland seems to support the differentiation of mucous neck to zymogenic cells. At the same time, high concentrations of the secreted SHH ligand seem to restrict the proliferation of surface pit cells [139, 140].
As SHH plays a crucially important role in cellular differentiation and gastric tissue homeostasis [141], epithelial cell differentiation in H. pylori-associated gastritis in the Mongolian gerbil model has been investigated. Persistent infection with H. pylori induced inflammation of the antrum and corpus of the stomach, which was accompanied by a clear loss of SHH expression in parietal cells and mucous neck cells of the gastric fundic glands as monitored by quantitative real-time (RT)-PCR, in situ hybridization, immunoblotting and immunohistochemistry. This phenotype was associated with the loss of parietal cells and disturbed fundic gland cell differentiation [142] (Fig. 3a). A similar observation was made in human patients underlining a correlation between H. pylori infections and the HH signaling components SHH, SMO and GLI2 [143]. In comparison to healthy mucosa, expression of the intestine-specific transcription factor caudal type homeobox 2 (CDX2) negatively correlated with SHH expression in the corpus lesser curve of gastric cancer patients indicating that H. pylori might employ SHH expression in gastric atrophy and intestinal metaplasia during the development and/or progression of gastric carcinogenesis [144] (Fig. 3a). CDX2 exhibits an important role in the development and maintenance of the intestinal epithelium, but is frequently found in gastric cancer with controversially discussed functions. The expression of CDX2 in transgenic mouse models transformed the gastric mucosa into intestinal metaplastic mucosa and triggered gastric cancer [145, 146], but in other studies CDX2 expression correlated with a better prognosis [147, 148]. Interestingly, H. pylori eradication led to an increase in SHH expression in Mongolian gerbils [149] and in the human corpus, where it mediated a decrease in CDX2 expression in the corpus lesser curve [150–152]. Although eradication of H. pylori mediated an increase in SHH expression and its downstream regulators, the beneficial effect was not observed in patients with high risk of gastric cancer [153]. Therefore, it was proposed that prevention of cancer might be improved through H. pylori eradication prior to the development of atrophic gastritis with intestinal metaplasia [153]. These observations also point to a functional role of SHH re-expression in the gastric epithelial regeneration. Notably, mice with a parietal cell-specific deletion of Shh (PC-Shh KO) showed a delayed wound healing [154], suggesting that SHH re-expression after H. pylori eradication contributes to epithelial regeneration.

Model of H. pylori interference with the SHH signaling pathway. a Infection with H. pylori induces a loss of SHH-expressing parietal cells (1). Loss of SHH might involve H. pylori-induced CDX2 expression, which then binds the SHH gene promoter leading to SHH downregulation (2) and to a dysfunction of parietal cells (8). Downregulation of SHH has also been associated with hypermethylation of the promoters of hedgehog signaling genes (3). The negative effect of H. pylori on SHH expression involves NF-κB activity (4) that induces the expression of cytokines to reduce SHH gene expression (5). On the other hand, SHH can be upregulated in response to calcium and PKC activity (11), which leads to the activation of GLI expression to enhance schlafen-4 (6) or increases H+/K+-ATPase gene expression (10). A positive effect of SHH on cytokine expression was observed (7), which could contribute to the NF-κB responses. H. pylori-induced proinflammatory cytokines further the dysfunction of parietal cells to inhibit gastric acid production (9), while an increased SHH expression promotes anti-apoptotic effect and proliferation (12). b H. pylori-increased SHH can function as a chemoattractant for macrophages and BM-MSCs. IFNγ-induced MSC proliferation required SHH secretion via an autocrine regulatory mechanism and appears to be involved in the repopulation of the inflamed tissue
H. pylori can directly control SHH expression
The loss of parietal cells associated with the loss of SHH expression during H. pylori-mediated atrophy suggests an indirect regulatory effect of H. pylori on HH activity. However, H. pylori may exert strategies directly targeting SHH expression. A comprehensive profiling of DNA methylation of a well-characterized series of primary gastric cancers was performed and 147 genes were identified exhibiting significantly changed methylation patterns in tumor and matched tumor-adjacent gastric tissue. Among these genes, hoxA5 and hedgehog signaling molecules (WNT2, WNT5A, SMO, HHIP, GLI3, BMP6) were found [155], suggesting that the loss of hedgehog signaling proteins is also the consequence of epigenetic processes (Fig. 3a). However, the reciprocal expression of SHH and CDX2 in intestinal metaplasia could not be explained by methylation [156]. SHH was drastically down-regulated in a CDX2 transgenic mouse model, which was not mediated by SHH promoter hypermethylation. In fact, the authors observed that CDX2 directly binds the TATA box in the SHH promoter leading to a down-regulation of the SHH expression, suggesting a direct connection between SHH and CDX2 expression. These observations were confirmed in cultured AGS, MKN45, and MKN74 cells transfected with CDX2-expressing plasmids, in which SHH expression was clearly decreased [156]. Although a cell-type-specific induction of CDX2 expression has been shown in H. pylori-infected gastric epithelial cell lines [157, 158], this observation has not yet been correlated with SHH gene regulation.
In insulin-gastrin (InsGas) mice that overexpress pancreatic gastrin to study gastric cancer, SHH, GLI1, but not GLI3 expression in pre-metaplastic lesions of non-infected mice was considerably reduced compared to normal adjacent glands, but partially re-expressed in H. felis-induced gastric metaplasia. It was suggested that H. felis-activated NF-κB and subsequently IL-8 secretion may be involved in this pathway [159] (Fig. 3a). This indicates that H. felis could regulate SHH signaling through the loss of SHH expressing cell types. Similar effects were found in transgenic GLI1-deficient mice infected with H. felis, which were largely resistant to the development of gastric metaplasia and infiltration by inflammatory cells [160]. GLI1 deletion blocked Th1 and Th2 cytokines, but not a Th17 response. As a target gene of GLI1, schlafen-4 was identified in microarray analyses (Fig. 3a), which was induced in wildtype mice, but not in the Gli1-deficient mice [160]. In addition, SHH has been shown to positively regulate cytokine expression during H. pylori infection [161]. Comparing WT and PC-Shh KO mice, an increase of Il-12, Il-1β, Il-10, Ifnγ and Mip-2 expression was only observed in wildtype mice infected with H. pylori [161] (Fig. 3a).
Although it was previously hypothesized that loss of SHH expression is mainly caused by the loss of parietal cells, it was further shown that downregulation of SHH expression is associated with parietal cell dysfunction revealing an important role in gastric acid secretion [162] (Fig. 3a). This might be supported by the finding that the H. pylori-induced proinflammatory cytokine IL-1β inhibited gastric acid production, intracellular calcium release, and SHH expression in parietal cells via IL-1 receptor signaling leading to gastric atrophy [163] (Fig. 3a). These data underline the importance of the acidic environment in maintaining SHH expression and secretion in the human stomach.
The interference of SHH and gastric acid became more complex when another report indicated that SHH can increase acid secretion by gastric parietal cells through an increase of H+/K+-ATPase gene expression [164] (Fig. 3a). Phenotypically, transgenic mice that express the hedgehog inhibitor Hhip1 secreted less gastric acid resulting in hypochlorhydria. In these mice, somatostatin was decreased, gastrin gene expression was enhanced and Shh mRNA was down-regulated. Shh gene expression could be activated through an increase of intracellular calcium, which then activates calcium-specific protein kinase C alpha and beta (PKC-α, PKC-β) (Fig. 3a). Therefore, SHH could function as a ligand that transduces alterations of gastric acidity to the secretion of gastrin by G cells [140].
Overexpression of SHH in human gastric cancers has also been reported [165–167]. In agreement to this, Shh expression can be induced in H. pylori-colonized mice in an acid-independent manner [168, 169]. In these mice, H. pylori induced infiltration of CD4+ T cells and increased levels of IFNγ and Il-1β in the stomach after six months of infection [169]. Intriguingly, PC-Shh KO mice did not develop gastritis in response to H. pylori and did not display elevated CD4+ T cells. Macrophages are crucially important in the development of gastritis [42]. In PC-Shh KO mice, macrophages were not recruited to the position where ulceration was induced [154]. Interestingly, H. pylori-increased SHH mainly occurred in parietal cells of the fundic mucosa and can function as a chemoattractant for macrophages as shown in bone marrow chimera experiments [169] (Fig. 3b). Accordingly, an organoid culture system for the fundic region of the mouse stomach that contained SHH-expressing parietal cells was established to investigate H. pylori-mediated SHH signaling. H. pylori activated NF-κB, which induced SHH expression in a CagA-dependent manner. Consequently, pharmacological inhibition of NF-κB blocked SHH upregulation [168]. From these data, the authors concluded that SHH acts as a regulator of the initial immune response. Underlining this assumption, CagA-positive H. pylori strains were reported to activate SHH expression in the cultured gastric epithelial cell lines AGS, MKN-28, MKN-45 and Kato III cells. Besides SHH, PTCH and GLI were upregulated as well. The authors suggested that H. pylori induced NF-κB activity in a CagA-dependent manner to activate SHH expression [170]. However, how CagA is implicated in NF-κB-associated SHH regulation needs to be investigated in more detail, since it is well established that CagA is not directly involved in early H. pylori-mediated NF-κB activation [33, 34]. Functionally, expression of SHH in cell culture experiments led to a higher resistance to apoptosis upon infection with H. pylori [171], which could explain the hyperproliferative phenotype in response to H. pylori infections.
HH/GLI signaling in the recruitment of bone-marrow derived mesenchymal stem cells (BM-MSCs) to inflamed tissues in response to H. pylori
SHH appears to be not only a potential chemoattractant for macrophages, but also for BM-MSCs in chronic inflammation [169, 172]. During chronic infection with H. pylori, BM-MSCs are recruited to the site of chronic inflammation to repopulate the gastric epithelium and promote gastric cancer progression [173]. Hence, investigations were performed to elucidate the role of SHH in the regulation of BM-MSCs in the stomach [174]. It was found that IFNγ-induced mesenchymal stem cell (MSC) proliferation required SHH secretion via an autocrine regulatory mechanism. Only MSCs that expressed SHH were finally recruited to the gastric mucosa in response to IFNγ [174] (Fig. 3b). Whether H. pylori activated T-lymphocytes produce IFNγ to trigger MSCs in the bone marrow to secrete elevated levels of SHH needs to be investigated in future experiments.
Gastritis can result in MSC proliferation as well. Using a gastrin-deficient mouse model exhibiting a hypochlorhydric phenotype leading to inflammation, parietal cell atrophy and metaplasia, BM-MSCs showed aberrant proliferation and activation of HH/GLI signaling in response to chronic gastric inflammation [175]. Parabiosis experiments demonstrated that circulating signals (e.g. TGFβ) released during H. pylori-mediated gastritis induced HH/GLI signaling within bone marrow-derived stromal cells and the rapid recruitment of MSCs to the inflamed stomach [175] (Fig. 3b).
The implication of hedgehog signaling in MSC recruitment is interesting and led to the question about the functional consequences of the recruitment of MSCs to inflamed tissue: tissue regeneration and/or gastric cancer? The finding that H. pylori can recruit MSCs that repopulate the epithelium and then transdifferentiate into intraepithelial cancer cells prompted the hypothesis that gastric epithelial cancer can originate from bone marrow-derived cells [173].
Conclusions
H. pylori infections are a paradigm for inflammation-driven cancer. A vast number of reports exist describing the pathophysiological mechanisms, though our knowledge of H. pylori-modulated hedgehog signaling in gastric homeostasis and malignant disease is still scarce. At a first glance, the influence of H. pylori on SHH expression and function appears controversial. However, upon a closer view on the complex processes it becomes apparently clear that a precise regulation of SHH is a crucial part of gastric physiology. Future studies are necessary to elucidate how gastric HH/GLI signaling is implicated in H. pylori-induced pathogenesis as pharmacological targeting of HH/GLI elements represents an attractive approach for the establishment of novel strategies for the treatment of gastric cancer.
Abbreviations
- Abl:
-
Abelson leukemia virus oncoprotein
- APC:
-
Adenomatosis polyposis coli
- BabA:
-
Blood group antigen-binding adhesin A
- BM-MSC:
-
Bone-marrow derived mesenchymal stem cells
- BMP:
-
Bone morphogenetic protein
- BMP6:
-
Bone morphogenetic protein 6
- BOC:
-
Brother of CDO
- CAF:
-
Cancer-associated fibroblasts
- CagA:
-
Cytotoxin-associated gene A
- CagPAI:
-
Cytotoxin-associated gene pathogenicity island
- cAMP:
-
Cyclic adenosine monophosphate
- CD4+ T cells:
-
CD4 positive T cells
- Cdh1:
-
E-cadherin
- CDO:
-
Cell adhesion molecule-related/down-regulated by oncogenes
- CDX2:
-
Caudal type homeobox 2
- CK1:
-
Casein kinase 1
- DLG5:
-
Disc large MAGUK scaffold protein 5
- ECL:
-
enterochromaffin-like cell
- GAS1:
-
Growth arrest specific 1
- GKO:
-
Gastrin knock-out
- GLI:
-
Glioblastoma-associated-protein
- GLI1:
-
Glioblastoma-associated-protein 1
- GLI2:
-
Glioblastoma-associated-protein 2
- GLI3:
-
Glioblastoma-associated-protein 3
- GLIA:
-
Glioblastoma-associated-protein activator form
- GLIR:
-
Glioblastoma-associated-protein repressor form
- GPR161:
-
G-protein-coupled receptor 161
- GSK3β:
-
Glycogen synthase kinase 3 beta
- H. felis :
-
Helicobacter felis
- H. pylori :
-
Helicobacter pylori
- H+/K+ ATPase:
-
Proton/Potassium exchanging ATPase
- HH:
-
Hedgehog
- HHIP:
-
Hedgehog interacting protein
- HOXA5:
-
Homeobox A5
- IFNγ:
-
Interferon gamma
- IFT:
-
Intraflagellar transport
- IL10:
-
Interleukin 10
- IL11:
-
Interleukin 11
- IL12:
-
Interleukin 12
- IL1B:
-
Interleukin 1 beta
- IL1RN:
-
Interleukin 1 receptor antagonist
- IL-8:
-
Interleukin 8
- InsGas:
-
Insulin-gastrin
- KIF7:
-
Kinesin family member 7
- KRAS:
-
Kirsten rat sarcoma viral oncogene homolog proto-oncogene
- MALT:
-
Mucosa-associated lymphoid tissue
- MIP-2:
-
Macrophage inflammatory protein 2 (aka CXCL2)
- MSC:
-
Mesenchymal stem cell
- NF-κB:
-
Nuclear factor kappa B
- PC-ShhKO :
-
Parietal cell-specific Shh knock-out
- PKA:
-
Protein kinase A
- PKC-α:
-
Protein kinase C alpha
- PKC-β:
-
Protein kinase C beta
- PTCH:
-
Patched
- RT-PCR:
-
Real time PCR
- SabA:
-
Sialic acid-binding adhesin A
- SHH:
-
Sonic hedgehog
- sHip-1:
-
Secreted form of HHIP
- SMO:
-
Smoothened
- Src:
-
Rous sarcoma oncogene
- STAT3:
-
Signal transducer and activator of transcription 3
- SUFU:
-
Suppressor of fused
- T4SS:
-
Type IV secretion system
- TAM:
-
Tumor-associated macrophages
- TGFβ:
-
Transforming growth factor beta
- TNF-alpha:
-
Tumor necrosis factor alpha
- TP53:
-
Tumor protein p53
- Tregs:
-
Regulatory T cells
- VacA:
-
Vacuolating cytotoxin A
- Wnt:
-
Wingless-type MMTV integration site family
- WNT2:
-
Wingless-type MMTV integration site family member 2
- WNT5A:
-
Wingless-type MMTV integration site family member 5 A
- WT:
-
Wildtype
References
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917.
Ekstrom AM, Held M, Hansson LE, Engstrand L, Nyren O. Helicobacter pylori in gastric cancer established by CagA immunoblot as a marker of past infection. Gastroenterology. 2001;121:784–91.
Peek Jr RM, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–48.
Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1-241.
Carcas LP. Gastric cancer review. J Carcinog. 2014;13:14.
Correa P. Gastric cancer: overview. Gastroenterol Clin North Am. 2013;42:211–7.
Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015;16:e60–70.
Fleisher AS, Esteller M, Wang S, Tamura G, Suzuki H, Yin J, Zou TT, Abraham JM, Kong D, Smolinski KN, Shi YQ, Rhyu MG, Powell SM, James SP, Wilson KT, Herman JG, Meltzer SJ. Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res. 1999;59:1090–5.
Camargo MC, Mera R, Correa P, Peek Jr RM, Fontham ET, Goodman KJ, Piazuelo MB, Sicinschi L, Zabaleta J, Schneider BG. Interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms and gastric cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;15:1674–87.
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni Jr JF, Rabkin CS. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402.
Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001;42:1337–48.
Krisch LM, Posselt G, Hammerl P, Wessler S. CagA Phosphorylation in Helicobacter pylori-Infected B Cells Is Mediated by the Nonreceptor Tyrosine Kinases of the Src and Abl Families. Infect Immun. 2016;84:2671–80.
Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest. 2012;122:1553–66.
Poppe M, Feller SM, Romer G, Wessler S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene. 2007;26:3462–72.
Backert S, Tegtmeyer N, Fischer W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015;10:955–65.
Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15:306–16.
Matos JI, de Sousa HA, Marcos-Pinto R, Dinis-Ribeiro M. Helicobacter pylori CagA and VacA genotypes and gastric phenotype: a meta-analysis. Eur J Gastroenterol Hepatol. 2013;25:1431–41.
Huang JQ, Zheng GF, Sumanac K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology. 2003;125:1636–44.
Franco AT, Johnston E, Krishna U, Yamaoka Y, Israel DA, Nagy TA, Wroblewski LE, Piazuelo MB, Correa P, Peek Jr RM. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–87.
Gaddy JA, Radin JN, Loh JT, Zhang F, Washington MK, Peek Jr RM, Algood HM, Cover TL. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infect Immun. 2013;81:2258–67.
Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, Yamada G, Azuma T, Hatakeyama M. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci U S A. 2008;105:1003–8.
Boquet P, Ricci V. Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol. 2012;20:165–74.
Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol. 2005;3:320–32.
Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesin. Proc Natl Acad Sci U S A. 1999;96:12778–83.
Yamaoka Y, Ojo O, Fujimoto S, Odenbreit S, Haas R, Gutierrez O, El-Zimaity HM, Reddy R, Arnqvist A, Graham DY. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut. 2006;55:775–81.
Posselt G, Backert S, Wessler S. The functional interplay of Helicobacter pylori factors with gastric epithelial cells induces a multi-step process in pathogenesis. Cell Commun Signal. 2013;11:77.
Schneider S, Carra G, Sahin U, Hoy B, Rieder G, Wessler S. Complex cellular responses of Helicobacter pylori-colonized gastric adenocarcinoma cells. Infect Immun. 2011;79:2362–71.
Wessler S, Backert S. Molecular mechanisms of epithelial-barrier disruption by Helicobacter pylori. Trends Microbiol. 2008;16:397–405.
Amieva M, Peek Jr RM. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology. 2016;150:64–78.
Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345:196–202.
Ishimoto T, Sawayama H, Sugihara H, Baba H. Interaction between gastric cancer stem cells and the tumor microenvironment. J Gastroenterol. 2014;49:1111–20.
Chung HW, Lim JB. Role of the tumor microenvironment in the pathogenesis of gastric carcinoma. World J Gastroenterol. 2014;20:1667–80.
Sokolova O, Borgmann M, Rieke C, Schweitzer K, Rothkotter HJ, Naumann M. Helicobacter pylori induces type 4 secretion system-dependent, but CagA-independent activation of IkappaBs and NF-kappaB/RelA at early time points. Int J Med Microbiol. 2013;303:548–52.
Schweitzer K, Sokolova O, Bozko PM, Naumann M. Helicobacter pylori induces NF-kappaB independent of CagA. EMBO Rep. 2010;11:10–1. author reply 11-12.
Aihara M, Tsuchimoto D, Takizawa H, Azuma A, Wakebe H, Ohmoto Y, Imagawa K, Kikuchi M, Mukaida N, Matsushima K. Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, MKN45. Infect Immun. 1997;65:3218–24.
Sharma SA, Tummuru MK, Blaser MJ, Kerr LD. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol. 1998;160:2401–7.
Beales IL, Calam J. Stimulation of IL-8 production in human gastric epithelial cells by Helicobacter pylori, IL-1beta and TNF-alpha requires tyrosine kinase activity, but not protein kinase C. Cytokine. 1997;9:514–20.
Suganuma M, Yamaguchi K, Ono Y, Matsumoto H, Hayashi T, Ogawa T, Imai K, Kuzuhara T, Nishizono A, Fujiki H. TNF-alpha-inducing protein, a carcinogenic factor secreted from H. pylori, enters gastric cancer cells. Int J Cancer. 2008;123:117–22.
Ernst M, Najdovska M, Grail D, Lundgren-May T, Buchert M, Tye H, Matthews VB, Armes J, Bhathal PS, Hughes NR, Marcusson EG, Karras JG, Na S, Sedgwick JD, Hertzog PJ, Jenkins BJ. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest. 2008;118:1727–38.
Merchant JL. What lurks beneath: IL-11, via Stat3, promotes inflammation-associated gastric tumorigenesis. J Clin Invest. 2008;118:1628–31.
Ohta M, Kitadai Y, Tanaka S, Yoshihara M, Yasui W, Mukaida N, Haruma K, Chayama K. Monocyte chemoattractant protein-1 expression correlates with macrophage infiltration and tumor vascularity in human gastric carcinomas. Int J Oncol. 2003;22:773–8.
Kaparakis M, Walduck AK, Price JD, Pedersen JS, van Rooijen N, Pearse MJ, Wijburg OL, Strugnell RA. Macrophages are mediators of gastritis in acute Helicobacter pylori infection in C57BL/6 mice. Infect Immun. 2008;76:2235–9.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6.
Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–54.
Kindlund B, Sjoling A, Hansson M, Edebo A, Hansson LE, Sjovall H, Svennerholm AM, Lundin BS. FOXP3-expressing CD4(+) T-cell numbers increase in areas of duodenal gastric metaplasia and are associated to CD4(+) T-cell aggregates in the duodenum of Helicobacter pylori-infected duodenal ulcer patients. Helicobacter. 2009;14:192–201.
Shen LS, Wang J, Shen DF, Yuan XL, Dong P, Li MX, Xue J, Zhang FM, Ge HL, Xu D. CD4(+)CD25(+)CD127(low/-) regulatory T cells express Foxp3 and suppress effector T cell proliferation and contribute to gastric cancers progression. Clin Immunol. 2009;131:109–18.
Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, Fujii H. Localisation pattern of Foxp3+ regulatory T cells is associated with clinical behaviour in gastric cancer. Br J Cancer. 2008;98:148–53.
Mizukami Y, Kono K, Kawaguchi Y, Akaike H, Kamimura K, Sugai H, Fujii H. CCL17 and CCL22 chemokines within tumor microenvironment are related to accumulation of Foxp3+ regulatory T cells in gastric cancer. Int J Cancer. 2008;122:2286–93.
Ichihara F, Kono K, Takahashi A, Kawaida H, Sugai H, Fujii H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin Cancer Res. 2003;9:4404–8.
Semba S, Kodama Y, Ohnuma K, Mizuuchi E, Masuda R, Yashiro M, Hirakawa K, Yokozaki H. Direct cancer-stromal interaction increases fibroblast proliferation and enhances invasive properties of scirrhous-type gastric carcinoma cells. Br J Cancer. 2009;101:1365–73.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401.
Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–7.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48.
Schubert ML. Functional anatomy and physiology of gastric secretion. Curr Opin Gastroenterol. 2015;31:479–85.
Schubert ML. Gastric secretion. Curr Opin Gastroenterol. 2014;30:578–82.
Kaneko H, Konagaya T, Kusugami K. Helicobacter pylori and gut hormones. J Gastroenterol. 2002;37:77–86.
De Datta D, Roychoudhury S. To be or not to be: The host genetic factor and beyond in Helicobacter pylori mediated gastro-duodenal diseases. World J Gastroenterol. 2015;21:2883–95.
Naumann M, Sokolova O, Tegtmeyer N, Backert S. Helicobacter pylori: A Paradigm Pathogen for Subverting Host Cell Signal Transmission. Trends Microbiol. 2017;25:316–28.
Fischer W, Prassl S, Haas R. Virulence mechanisms and persistence strategies of the human gastric pathogen Helicobacter pylori. Curr Top Microbiol Immunol. 2009;337:129–71.
Wu WK, Cho CH, Lee CW, Fan D, Wu K, Yu J, Sung JJ. Dysregulation of cellular signaling in gastric cancer. Cancer Lett. 2010;295:144–53.
Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801.
Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007;17:438–47.
Teglund S, Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 1805;2010:181–208.
Kasper M, Regl G, Frischauf AM, Aberger F. GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur J Cancer. 2006;42:437–45.
Basset-Seguin N, Sharpe HJ, de Sauvage FJ. Efficacy of Hedgehog pathway inhibitors in Basal cell carcinoma. Mol Cancer Ther. 2015;14:633–41.
Atwood SX, Chang AL, Oro AE. Hedgehog pathway inhibition and the race against tumor evolution. J Cell Biol. 2012;199:193–7.
Price MA, Kalderon D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell. 2002;108:823–35.
Tempe D, Casas M, Karaz S, Blanchet-Tournier MF, Concordet JP. Multisite protein kinase A and glycogen synthase kinase 3beta phosphorylation leads to Gli3 ubiquitination by SCFbetaTrCP. Mol Cell Biol. 2006;26:4316–26.
Wen X, Lai CK, Evangelista M, Hongo JA, de Sauvage FJ, Scales SJ. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol Cell Biol. 2010;30:1910–22.
Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–53.
Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–34.
Goodrich LV, Scott MP. Hedgehog and patched in neural development and disease. Neuron. 1998;21:1243–57.
Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–17.
Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–72.
Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet. 2011;12:393–406.
Wong SY, Reiter JF. The primary cilium at the crossroads of mammalian hedgehog signaling. Curr Top Dev Biol. 2008;85:225–60.
Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–29.
Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–37.
Tenzen T, Allen BL, Cole F, Kang JS, Krauss RS, McMahon AP. The cell surface membrane proteins Cdo and Boc are components and targets of the Hedgehog signaling pathway and feedback network in mice. Dev Cell. 2006;10:647–56.
Allen BL, Tenzen T, McMahon AP. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007;21:1244–57.
McLellan JS, Zheng X, Hauk G, Ghirlando R, Beachy PA, Leahy DJ. The mode of Hedgehog binding to Ihog homologues is not conserved across different phyla. Nature. 2008;455:979–83.
Chuang PT, McMahon AP. Vertebrate Hedgehog signalling modulated by induction of a Hedgehog-binding protein. Nature. 1999;397:617–21.
Milenkovic L, Scott MP, Rohatgi R. Lateral transport of Smoothened from the plasma membrane to the membrane of the cilium. J Cell Biol. 2009;187:365–74.
Chong YC, Mann RK, Zhao C, Kato M, Beachy PA. Bifurcating action of Smoothened in Hedgehog signaling is mediated by Dlg5. Genes Dev. 2015;29:262–76.
Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, Jackson PK. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 2013;152:210–23.
Mukhopadhyay S, Rohatgi R. G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin Cell Dev Biol. 2014;33:63–72.
Byrne EF, Sircar R, Miller PS, Hedger G, Luchetti G, Nachtergaele S, Tully MD, Mydock-McGrane L, Covey DF, Rambo RP, Sansom MS, Newstead S, Rohatgi R, Siebold C. Structural basis of Smoothened regulation by its extracellular domains. Nature. 2016;535:517–22.
Luchetti G, Sircar R, Kong JH, Nachtergaele S, Sagner A, Byrne EF, Covey DF, Siebold C, Rohatgi R. Cholesterol activates the G-protein coupled receptor Smoothened to promote Hedgehog signaling. Elife. 2016;5:e20304.
Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci U S A. 2009;106:21666–71.
Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191:415–28.
Ikram MS, Neill GW, Regl G, Eichberger T, Frischauf AM, Aberger F, Quinn A, Philpott M. GLI2 is expressed in normal human epidermis and BCC and induces GLI1 expression by binding to its promoter. J Invest Dermatol. 2004;122:1503–9.
Regl G, Neill GW, Eichberger T, Kasper M, Ikram MS, Koller J, Hintner H, Quinn AG, Frischauf AM, Aberger F. Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene. 2002;21:5529–39.
Callahan CA, Ofstad T, Horng L, Wang JK, Zhen HH, Coulombe PA, Oro AE. MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 2004;18:2724–9.
Huntzicker EG, Estay IS, Zhen H, Lokteva LA, Jackson PK, Oro AE. Dual degradation signals control Gli protein stability and tumor formation. Genes Dev. 2006;20:276–81.
Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013;494:484–8.
Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, Pietrosanti L, De Smaele E, Ferretti E, Miele E, Pelloni M, De Simone G, Pedone EM, Gallinari P, Giorgi A, Steinkuhler C, Vitagliano L, Pedone C, Schinin ME, Screpanti I, Gulino A. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132–42.
Coni S, Antonucci L, D’Amico D, Di Magno L, Infante P, De Smaele E, Giannini G, Di Marcotullio L, Screpanti I, Gulino A, Canettieri G. Gli2 acetylation at lysine 757 regulates hedgehog-dependent transcriptional output by preventing its promoter occupancy. PLoS One. 2013;8:e65718.
Di Marcotullio L, Ferretti E, Greco A, De Smaele E, Po A, Sico MA, Alimandi M, Giannini G, Maroder M, Screpanti I, Gulino A. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat Cell Biol. 2006;8:1415–23.
Winklmayr M, Schmid C, Laner-Plamberger S, Kaser A, Aberger F, Eichberger T, Frischauf AM. Non-consensus GLI binding sites in Hedgehog target gene regulation. BMC Mol Biol. 2010;11:2.
Balaskas N, Ribeiro A, Panovska J, Dessaud E, Sasai N, Page KM, Briscoe J, Ribes V. Gene regulatory logic for reading the Sonic Hedgehog signaling gradient in the vertebrate neural tube. Cell. 2012;148:273–84.
Cohen M, Briscoe J, Blassberg R. Morphogen interpretation: the transcriptional logic of neural tube patterning. Curr Opin Genet Dev. 2013;23:423–8.
Aberger F, Ruiz IAA. Context-dependent signal integration by the GLI code: the oncogenic load, pathways, modifiers and implications for cancer therapy. Semin Cell Dev Biol. 2014;33:93–104.
Riobo NA, Haines GM, Emerson Jr CP. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006;66:839–45.
Riobo NA, Lu K, Ai X, Haines GM, Emerson Jr CP. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc Natl Acad Sci U S A. 2006;103:4505–10.
Teperino R, Aberger F, Esterbauer H, Riobo N, Pospisilik JA. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Semin Cell Dev Biol. 2014;33:81–92.
Lauth M, Bergstrom A, Toftgard R. Phorbol esters inhibit the Hedgehog signalling pathway downstream of Suppressor of Fused, but upstream of Gli. Oncogene. 2007;26:5163–8.
Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F, Ruiz IAA. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007;104:5895–900.
Eberl M, Klingler S, Mangelberger D, Loipetzberger A, Damhofer H, Zoidl K, Schnidar H, Hache H, Bauer HC, Solca F, Hauser-Kronberger C, Ermilov AN, Verhaegen ME, Bichakjian CK, Dlugosz AA, Nietfeld W, Sibilia M, Lehrach H, Wierling C, Aberger F. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol Med. 2012;4:218–33.
Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, Aberger F. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009;69:1284–92.
Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, Schmid C, Hauser-Kronberger C, Regl G, Philpott MP, Aberger F. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol. 2006;26:6283–98.
Stecca B, Ruiz IAA. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J Mol Cell Biol. 2010;2:84–95.
Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013;19:1410–22.
Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8.
Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Mackey HM, Lum BL, Darbonne WC, Marsters Jr JC, de Sauvage FJ, Low JA. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–72.
Tang JY, Mackay-Wiggan JM, Aszterbaum M, Yauch RL, Lindgren J, Chang K, Coppola C, Chanana AM, Marji J, Bickers DR, Epstein Jr EH. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N Engl J Med. 2012;366:2180–8.
Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang AL, Low JA, Mackey HM, Yauch RL, Graham RA, Reddy JC, Hauschild A. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171–9.
Sekulic A, Von Hoff D. Hedgehog Pathway Inhibition. Cell. 2016;164:831.
Ng JM, Curran T. The Hedgehog’s tale: developing strategies for targeting cancer. Nat Rev Cancer. 2011;11:493–501.
Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–51.
Karhadkar SS, Bova GS, Abdallah N, Dhara S, Gardner D, Maitra A, Isaacs JT, Berman DM, Beachy PA. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431:707–12.
Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–7.
Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, Ruiz i Altaba A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO molecular medicine. 2009;1:338–51.
Varnat F, Zacchetti G, Ruiz i Altaba A. Hedgehog pathway activity is required for the lethality and intestinal phenotypes of mice with hyperactive Wnt signaling. Mech Dev. 2010;127:73–81.
Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, Nannini-Pepe M, Kotkow K, Marsters JC, Rubin LL, de Sauvage FJ. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–10.
Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30:303–12.
Mueller MT, Hermann PC, Witthauer J, Rubio-Viqueira B, Leicht SF, Huber S, Ellwart JW, Mustafa M, Bartenstein P, D’Haese JG, Schoenberg MH, Berger F, Jauch KW, Hidalgo M, Heeschen C. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology. 2009;137:1102–13.
Lee JJ, Perera RM, Wang H, Wu DC, Liu XS, Han S, Fitamant J, Jones PD, Ghanta KS, Kawano S, Nagle JM, Deshpande V, Boucher Y, Kato T, Chen JK, Willmann JK, Bardeesy N, Beachy PA. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A. 2014;111:E3091–100.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, Westphalen CB, Kitajewski J, Fernandez-Barrena MG, Fernandez-Zapico ME, Iacobuzio-Donahue C, Olive KP, Stanger BZ. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47.
Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ, de Sauvage FJ. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106:4254–9.
Nolan-Stevaux O, Lau J, Truitt ML, Chu GC, Hebrok M, Fernandez-Zapico ME, Hanahan D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23:24–36.
Rajurkar M, De Jesus-Monge WE, Driscoll DR, Appleman VA, Huang H, Cotton JL, Klimstra DS, Zhu LJ, Simin K, Xu L, McMahon AP, Lewis BC, Mao J. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc Natl Acad Sci U S A. 2012;109:E1038–47.
Gerling M, Buller NV, Kirn LM, Joost S, Frings O, Englert B, Bergstrom A, Kuiper RV, Blaas L, Wielenga MC, Almer S, Kuhl AA, Fredlund E, van den Brink GR, Toftgard R. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat Commun. 2016;7:12321.
Lee JJ, Rothenberg ME, Seeley ES, Zimdahl B, Kawano S, Lu WJ, Shin K, Sakata-Kato T, Chen JK, Diehn M, Clarke MF, Beachy PA. Control of inflammation by stromal Hedgehog pathway activation restrains colitis. Proc Natl Acad Sci U S A. 2016;113:E7545–53.
Merchant JL. Hedgehog signalling in gut development, physiology and cancer. J Physiol. 2012;590:421–32.
van den Brink GR. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol Rev. 2007;87:1343–75.
McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114.
Kim JH, Huang Z, Mo R. Gli3 null mice display glandular overgrowth of the developing stomach. Dev Dyn. 2005;234:984–91.
Engevik AC, Feng R, Yang L, Zavros Y. The acid-secreting parietal cell as an endocrine source of Sonic Hedgehog during gastric repair. Endocrinology. 2013;154:4627–39.
Xiao C, Ogle SA, Schumacher MA, Orr-Asman MA, Miller ML, Lertkowit N, Varro A, Hollande F, Zavros Y. Loss of parietal cell expression of Sonic hedgehog induces hypergastrinemia and hyperproliferation of surface mucous cells. Gastroenterology. 2010;138:550–61. 561 e551-558.
El-Zaatari M, Zavros Y, Tessier A, Waghray M, Lentz S, Gumucio D, Todisco A, Merchant JL. Intracellular calcium release and protein kinase C activation stimulate sonic hedgehog gene expression during gastric acid secretion. Gastroenterology. 2010;139:2061–71. e2062.
Katoh Y, Katoh M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review). Int J Mol Med. 2006;18:1019–23.
Suzuki H, Minegishi Y, Nomoto Y, Ota T, Masaoka T, van den Brink GR, Hibi T. Down-regulation of a morphogen (sonic hedgehog) gradient in the gastric epithelium of Helicobacter pylori-infected Mongolian gerbils. J Pathol. 2005;206:186–97.
Stasikowska-Kanicka O, Wagrowska-Danilewicz M, Bialek I, Danilewicz M. The immunoexpression of Shh, Smo and Gli2 in Helicobacter pylori positive and negative gastric biopsies. Pol J Pathol. 2012;63:25–30.
Shiotani A, Iishi H, Uedo N, Ishihara R, Ishiguro S, Tatsuta M, Nakae Y, Kumamoto M, Hinoi T, Merchant JL. Helicobacter pylori-induced atrophic gastritis progressing to gastric cancer exhibits sonic hedgehog loss and aberrant CDX2 expression. Aliment Pharmacol Ther. 2006;24 Suppl 4:71–80.
Silberg DG, Sullivan J, Kang E, Swain GP, Moffett J, Sund NJ, Sackett SD, Kaestner KH. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology. 2002;122:689–96.
Mutoh H, Sakurai S, Satoh K, Tamada K, Kita H, Osawa H, Tomiyama T, Sato Y, Yamamoto H, Isoda N, Yoshida T, Ido K, Sugano K. Development of gastric carcinoma from intestinal metaplasia in Cdx2-transgenic mice. Cancer Res. 2004;64:7740–7.
Fan Z, Li J, Dong B, Huang X. Expression of Cdx2 and hepatocyte antigen in gastric carcinoma: correlation with histologic type and implications for prognosis. Clin Cancer Res. 2005;11:6162–70.
Roessler K, Monig SP, Schneider PM, Hanisch FG, Landsberg S, Thiele J, Holscher AH, Dienes HP, Baldus SE. Co-expression of CDX2 and MUC2 in gastric carcinomas: correlations with clinico-pathological parameters and prognosis. World J Gastroenterol. 2005;11:3182–8.
Nishizawa T, Suzuki H, Nakagawa I, Minegishi Y, Masaoka T, Iwasaki E, Hibi T. Early Helicobacter pylori eradication restores sonic hedgehog expression in the gastric mucosa of Mongolian gerbils. Digestion. 2009;79:99–108. doi:10.1159/000209219. Epub 000202009 Mar 000209218.
Shiotani A, Uedo N, Iishi H, Tatsuta M, Ishiguro S, Nakae Y, Kamada T, Haruma K, Merchant JL. Re-expression of sonic hedgehog and reduction of CDX2 after Helicobacter pylori eradication prior to incomplete intestinal metaplasia. Int J Cancer. 2007;121:1182–9.
Nishizawa T, Suzuki H, Masaoka T, Minegishi Y, Iwasahi E, Hibi T. Helicobacter pylori eradication restored sonic hedgehog expression in the stomach. Hepatogastroenterology. 2007;54:697–700.
Jung DH, Kim JH, Lee YC, Lee SK, Shin SK, Park JC, Chung HS, Kim H, Kim H, Kim YH, Park JJ, Youn YH, Park H. Helicobacter pylori Eradication Reduces the Metachronous Recurrence of Gastric Neoplasms by Attenuating the Precancerous Process. J Gastric Cancer. 2015;15:246–55.
Shiotani A, Murao T, Uedo N, Iishi H, Yamanaka Y, Kamada T, Kusunoki H, Inoue K, Haruma K. Eradication of H. pylori did not improve abnormal sonic hedgehog expression in the high risk group for gastric cancer. Dig Dis Sci. 2012;57:643–9.
Xiao C, Feng R, Engevik AC, Martin JR, Tritschler JA, Schumacher M, Koncar R, Roland J, Nam KT, Goldenring JR, Zavros Y. Sonic Hedgehog contributes to gastric mucosal restitution after injury. Lab Invest. 2013;93:96–111.
Loh M, Liem N, Vaithilingam A, Lim PL, Sapari NS, Elahi E, Mok ZY, Cheng CL, Yan B, Pang B, Salto-Tellez M, Yong WP, Iacopetta B, Soong R. DNA methylation subgroups and the CpG island methylator phenotype in gastric cancer: a comprehensive profiling approach. BMC Gastroenterol. 2014;14:55.
Mutoh H, Hayakawa H, Sashikawa M, Sakamoto H, Sugano K. Direct repression of Sonic Hedgehog expression in the stomach by Cdx2 leads to intestinal transformation. Biochem J. 2010;427:423–34.
Barros R, Marcos N, Reis CA, De Luca A, David L, Almeida R. CDX2 expression is induced by Helicobacter pylori in AGS cells. Scand J Gastroenterol. 2009;44:124–5.
Matsuda K, Yamauchi K, Matsumoto T, Sano K, Yamaoka Y, Ota H. Quantitative analysis of the effect of Helicobacter pylori on the expressions of SOX2, CDX2, MUC2, MUC5AC, MUC6, TFF1, TFF2, and TFF3 mRNAs in human gastric carcinoma cells. Scand J Gastroenterol. 2008;43:25–33.
El-Zaatari M, Tobias A, Grabowska AM, Kumari R, Scotting PJ, Kaye P, Atherton J, Clarke PA, Powe DG, Watson SA. De-regulation of the sonic hedgehog pathway in the InsGas mouse model of gastric carcinogenesis. Br J Cancer. 2007;96:1855–61.
El-Zaatari M, Kao JY, Tessier A, Bai L, Hayes MM, Fontaine C, Eaton KA, Merchant JL. Gli1 deletion prevents Helicobacter-induced gastric metaplasia and expansion of myeloid cell subsets. PLoS One. 2013;8:e58935.
Marwaha S, Schumacher MA, Zavros Y, Eghbalnia HR. Crosstalks between cytokines and Sonic Hedgehog in Helicobacter pylori infection: a mathematical model. PLoS One. 2014;9:e111338.
Minegishi Y, Suzuki H, Arakawa M, Fukushima Y, Masaoka T, Ishikawa T, Wright NA, Hibi T. Reduced Shh expression in TFF2-overexpressing lesions of the gastric fundus under hypochlorhydric conditions. J Pathol. 2007;213:161–9.
Waghray M, Zavros Y, Saqui-Salces M, El-Zaatari M, Alamelumangapuram CB, Todisco A, Eaton KA, Merchant JL. Interleukin-1beta promotes gastric atrophy through suppression of Sonic Hedgehog. Gastroenterology. 2010;138:562–72. 572 e561-562.
Stepan V, Ramamoorthy S, Nitsche H, Zavros Y, Merchant JL, Todisco A. Regulation and function of the sonic hedgehog signal transduction pathway in isolated gastric parietal cells. J Biol Chem. 2005;280:15700–8.
Wang LH, Choi YL, Hua XY, Shin YK, Song YJ, Youn SJ, Yun HY, Park SM, Kim WJ, Kim HJ, Choi JS, Kim SH. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Mod Pathol. 2006;19:675–83.
Fukaya M, Isohata N, Ohta H, Aoyagi K, Ochiya T, Saeki N, Yanagihara K, Nakanishi Y, Taniguchi H, Sakamoto H, Shimoda T, Nimura Y, Yoshida T, Sasaki H. Hedgehog signal activation in gastric pit cell and in diffuse-type gastric cancer. Gastroenterology. 2006;131:14–29.
Lee SY, Han HS, Lee KY, Hwang TS, Kim JH, Sung IK, Park HS, Jin CJ, Choi KW. Sonic hedgehog expression in gastric cancer and gastric adenoma. Oncol Rep. 2007;17:1051–5.
Schumacher MA, Feng R, Aihara E, Engevik AC, Montrose MH, Ottemann KM, Zavros Y. Helicobacter pylori-induced Sonic Hedgehog expression is regulated by NFkappaB pathway activation: the use of a novel in vitro model to study epithelial response to infection. Helicobacter. 2015;20:19–28.
Schumacher MA, Donnelly JM, Engevik AC, Xiao C, Yang L, Kenny S, Varro A, Hollande F, Samuelson LC, Zavros Y. Gastric Sonic Hedgehog acts as a macrophage chemoattractant during the immune response to Helicobacter pylori. Gastroenterology. 2012;142:1150–9. e1156.
Kim JH, Choi YJ, Lee SH, Shin HS, Lee IO, Kim YJ, Kim H, Yang WI, Kim H, Lee YC. Effect of Helicobacter pylori infection on the sonic hedgehog signaling pathway in gastric cancer cells. Oncol Rep. 2010;23:1523–8.
Lee KM, Lee JS, Jung HS, Park DK, Park HS, Hahm KB. Late reactivation of sonic hedgehog by Helicobacter pylori results in population of gastric epithelial cells that are resistant to apoptosis: implication for gastric carcinogenesis. Cancer Lett. 2010;287:44–53.
Warzecha J, Gottig S, Bruning C, Lindhorst E, Arabmothlagh M, Kurth A. Sonic hedgehog protein promotes proliferation and chondrogenic differentiation of bone marrow-derived mesenchymal stem cells in vitro. J Orthop Sci. 2006;11:491–6.
Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–71.
Donnelly JM, Chawla A, Houghton J, Zavros Y. Sonic hedgehog mediates the proliferation and recruitment of transformed mesenchymal stem cells to the stomach. PLoS One. 2013;8:e75225.
Donnelly JM, Engevik AC, Engevik M, Schumacher MA, Xiao C, Yang L, Worrell RT, Zavros Y. Gastritis promotes an activated bone marrow-derived mesenchymal stem cell with a phenotype reminiscent of a cancer-promoting cell. Dig Dis Sci. 2014;59:569–82.
Acknowledgements
Not applicable.
Funding
The work of S.W. was supported by the grants P24315 and W1213 from the Austrian Science Fund (FWF), work of F.A. by FWF grants P25629 and W1213 as well as by the priority program ACBN of the University of Salzburg and a cancer research grant from the County of Salzburg.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Authors’ contributions
SW, FA, LK and DE wrote the article. SW, LK and DE designed and assembled the figures. SW and FA conceived the content of the review and made final corrections and amendments. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wessler, S., Krisch, L.M., Elmer, D.P. et al. From inflammation to gastric cancer – the importance of Hedgehog/GLI signaling in Helicobacter pylori-induced chronic inflammatory and neoplastic diseases. Cell Commun Signal 15, 15 (2017). https://doi.org/10.1186/s12964-017-0171-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-017-0171-4