
Abstract
We propose Langerhans cell histiocytosis (LCH) is an inflammatory process that is prolonged by mutations. We hypothesize that Merkel cell polyomavirus (MCPyV) infection triggers an interleukin-1 (IL-1) activation loop that underlies the pathogenesis of LCH. Langerhans cells (LCs) are antigen presenting cells in the skin. When LCs encounter exogenous antigens, they migrate from the epidermis into draining lymphoid tissues to initiate T-cell activity. It has been proposed that LC migration-related factors, including E-cadherin, matrix metalloproteinase, and Notch ligand induce LCH activity. We found that the tyrosine phosphatase SHP-1, which binds IL-1 receptor-associated kinase 1, is expressed at a significantly higher level in LCH affecting multiple organ systems (MS-LCH) than in LCH affecting a single organ system (SS-LCH). IL-1 stimulates T helper 17 cells and their signature cytokine IL-17 had been a matter of controversy. We detected higher levels of IL-17A receptor expression in MS-LCH than in SS-LCH and proposed an IL-17 endocrine model that could settle the controversy. IL-1 is the first cytokine secreted in response to sensitizers and promotes LC migration from sentinel tissues. Myeloid differentiation primary response 88 (MyD88), downstream of the IL-1 receptor, has functions in both RAS signaling and inflammation, leading to human cell transformation. In 2010, an activating mutation in the B-rapidly accelerated fibrosarcoma gene (BRAF) V600E was found in LCH. This BRAF mutation induces phosphorylation of the extracellular signal-regulated kinase (ERK) that may play an important role with MyD88 in LCH pathogenesis. However, phosphorylated ERK (pERK) is rapidly dephosphorylated by dual specificity phosphatase 6 (DUSP6), and limited proliferation is predicted in BRAF mutant cells. MyD88 binds pERK via its D-domain, thereby preventing pERK–DUSP6 interaction and maintaining ERK in an active, phosphorylated state. We detected MCPyV-DNA in the peripheral blood cells of two out of three patients with LCH in high-risk organs but not in those of patients with LCH in non–high-risk organs (0/12; P = .029). MCPyV infection can trigger precursor LCH cells with BRAF mutation to produce IL-1; the IL-1 loop is amplified in all LCH subclasses. Our model indicates both BRAF mutation and IL-1 loop regulation as potential therapeutic targets.
Similar content being viewed by others
Introduction
Langerhans cell (LC) histiocytosis (LCH) is characterized by the proliferation of CD1a-positive abnormal LC-like cells (LCH cells). LCH is classified by its involvement of either a single organ system (SS-LCH) or multiple organ systems (MS-LCH) [1]. The latter form is frequent in children younger than 2 years, whereas SS-LCH is more common in children older than 2 years [2,3]. This rare disease affects 4–9 children per million each year [4-6]. The liver, spleen, and bone marrow (BM) are considered high-risk organs for LCH [7,8]. Therefore, LCH is also classified as involving at least one high-risk organ [LCH-risk organ (RO) (+)] or a no high-risk organ [LCH-RO (−)] [7]. Though most patients with LCH-RO (+) develop MS-LCH, some patients with the involvement of only one high-risk organ have a milder case, with symptoms similar to those observed in SS-LCH [9,10]. The morphology of lesions is so uniform that pathologists cannot determine whether a given biopsy originates from a patient with SS-LCH or MS-LCH, from a patient with LCH-RO (+) or LCH-RO (−), or from a child or an adult [11]. However, the clinical course of LCH is remarkably variable, ranging from lesions that spontaneously resolve, to a chronic disease that can be widespread and sometimes lethal [12-15].
Although LCH was first described a century ago, the etiology is still not understood [16]. For decades, it was thought that the disease is a reactive disorder rather than a neoplastic process [16]. As the former name, “eosinophilic granuloma” indicates that lesional LCH morphology is reminiscent of tissue reactions to an intracellular pathogen, of which tuberculous granuloma is the prototype [11]. Scabies infections are reported to induce LC hyperplasia, which mimics LCH [17]. However, recent studies indicate that LCH has a more neoplastic character [18-20]. While unexpected remission can rarely occur in neoplasms, spontaneous healing is more common in LCH, suggesting that there may be multiple pathobiologic contributions to the LCH process [11,21,22]. For example, an epidemiologic study revealed that risk factors for MS-LCH include an increase in infections, the use of antibiotics in the first 6 months of life, and a family history of thyroid disease, whereas SS-LCH is significantly associated with diarrhea and vomiting in the postnatal period [23].
In this review, we propose a new model for LCH pathogenesis in which the disease is a reactive disorder with underlying neoplastic potential. In other words, LCH is an inflammatory process that is prolonged by mutations.
Review
Langerhans cells
In 1868, Langerhans [24] described a new epidermal cell type with dendritic extensions, which he believed to be part of the skin neural network [25]. Later named Langerhans cells (LCs), these cells differ from the Merkel cells that were first described in 1875 by Merkel as touch cells [26] (Figures 1 and 2). The function of LCs was unknown until 1973, when Steinman et al. [27,28] first discovered that dendritic cells (DCs) are in fact antigen presenting cells.

Langerhans cells and Merkel cells in the epidermis. (A) Skin of one-year-old female baby with nevus cell nevus. (Hematoxylin and Eosin (H&E) stain). (B) Immunohistochemistry shows S100+ immature Langerhans cell (LC; red), nevus cell (NC; red), and cytokeratin 20 (CK20) + Merkel cells (MC; brown). Same area of panel A. (C) S100+ immature LC and NC (red) and CK20+ MC (brown) in a hair follicle. High power view of the left central part of panel B. (D) Immunohistochemistry shows CD1a + LC (blue) and CD1a- NC. Brown pigments correspond to melanin. Same area of panel A. (E) Immature LCs in the epidermis. High power view of panel D. (F) Immature LCs in a hair follicle are positive for CD1a, while the NCs are negative for CD1a. High power view of panel D.

Wild-type monocytes, wild-type LC precursors, or wild-type immature Langerhans cells without mutations. In these cells, mitogens such as growth factors bind to and activate cell-surface receptors (GFR: growth factor receptor) that induce signaling through a complex consisting of adaptor proteins and exchange factors to activate RAS (Blue circle) on the inner surface of the cell membrane. Once activated, RAS binds to and activates the RAF family of proteins, including BRAF, which subsequently phosphorylates and activates MEK. Activated MEK subsequently phosphorylates and activates ERK. Activated ERK phosphorylates numerous substrates within the cytoplasm and nucleus, promoting cell division and enhancing survival, movement, and differentiation. In the case of Merkel cell polyomavirus (MCPyV) infection, some LCs may present MCPyV antigen, inducing adaptive immunity through Toll-like receptors (TLRs). IL-1 is the first cytokine secreted in response to sensitizers. IL-1 binds to IL-1 receptor (IL-1R) and promotes LC migration from sentinel tissue such as the skin. MCPyV interferes with LC function and maturation to evade immune surveillance, which might allow infection by inhibiting NF-κB essential modulator (NEMO) and down-regulation of TLR9.
LCs are specialized immature DCs present in the skin (Figure 1). When LCs encounter exogenous antigens, they migrate from the epidermis to draining lymphoid tissues to initiate and present the major histocompatibility complex/peptide complexes to T cells. During this migration, LCs mature and the pattern of expression of their cell surface molecules changes [29,30]. LCs undergo the following steps for migration to the lymph nodes: down-regulation in the expression of E-cadherin, which anchors LCs to the epidermis; production of matrix metalloproteinase (MMP), which is required for passage through the basement membrane; an increase in the expression of the chemokine receptor CCR7, which guides migration toward CCL19 (MIP-3β) and CCL21 (SLC) [31]; and production of Notch ligands such as Delta and Jagged, which instruct distinct CD4 T helper cell fates [32].
LCs are normally generated and maintained locally in the steady state from precursors in the epidermis itself (Figure 3) [33]. This is sufficient to produce the low-level, steady-state efflux of LCs to the draining lymph nodes [33]. In mice, replenishment of LCs from bone-marrow precursors can only be observed experimentally following depletion of the LC population by local skin inflammation [33]. Then, the LC precursors are replenished by inflammatory monocytes that enter the epidermis from the bloodstream [33]. This ‘emergency’ replenishment of LCs might also be a model for the origin of LCs from non-inflammatory blood monocytes during early development, and perhaps a model for an ongoing, low-frequency event in the steady state [33].

Proposed relationship between MCPyV and wild-type Langerhans cell (LC) precursors and wild-type LCs or mutated monocytes and mutated LC precursors. (A) MCPyV usually causes inapparent infection with immunoglobulin production against MCPyV, indicating acquired immunity against MCPyV is actuated by antigen presenting cells. LCs are antigen presenting cells derived from bone marrow (BM). LCs are normally generated and maintained locally in the steady state from precursors in the epidermis itself. In inflammation LC precursors are replenished by monocytes. Monocytes and LC precursors are candidate reservoir cells for MCPyV. (B) On the contrary, mutated precursor LCH cells (mutant monocytes, mutant LC precursors or mutant LCs) do not show inapparent infection against MCPyV; in such cases, a reactive disorder might be triggered, such as proliferation of LCH cells, cytokine storms including IL-1β, and clinical remissions. Mutated precursor LCH cells (mutant monocytes) in blood vessels recognize MCPyV and induce LCH-RO (+). Mutated LCH precursor cells (mutant LC precursors or mutant LCS) in peripheral tissues recognize MCPyV and induce LCH-RO (−). Most patients with LCH-RO (+) develop MS-LCH. Some patients with only one high-risk organ involved have a milder clinical course, similar to that observed in SS-LCH. In addition to IL-1 loop, serum level of IL-18 (one of IL-1 agonists) and osteopontin that is closely related to IL-1 levels were significantly higher in LCH-RO (+) than that found in LCH-RO (−).
LCH activity is controlled by enzymes and cytokines that are involved in LC migration and antigen presentation [34-40]. LCH is characterized by a lesional cytokine storm, the prominent cytokine sources being both LCH cells and T cells [16]. Interleukin (IL)-1, IL-18, and tumor necrosis factor-alpha (TNF-α) are important cytokines that promote LC migration from the skin [41-43]. LCs respond to many chemokines, in particular CCL20, which appears to be the most powerful chemokine to induce migration [16,44]. During pathogen invasion, immature LCs expressing CCR6, the major functional CCL20 receptor, would be attracted to the site of inflammation. After antigen uptake, maturation of LCs results in downregulation of CCR6 and expression of CCR7, resulting in attraction to CCL19 and CCL21 which are expressed in the T-zones of lymph nodes [45]. Flemming et al. [46] reported coincident expression of both CCR6 and CCR7 by LCH cells. MMPs such as MMP-1 [47] and MMP-9 [48,49] are also important to migration of LCs and are expressed in LCH cells (GSE16395) [50]. LCs and LCH cells can be recognized using electron microscopy by the presence of specific Birbeck granules [1], and by immunohistochemical staining with antibodies recognizing langerin (CD207) or CD1a [51]. Immature LCs are phagocytic cells, and the calcium-dependent (C-type) lectin langerin (CD207) plays a role in antigen capture and subsequent Birbeck-granule formation [52].
Although some LCH cells have close relationship with leukemia/lymphoma cells [53,54], origins of LCH cells were not determined. Generally it is not easy to conclude cell origins of neoplastic cells even by immunoglobulin productions [55]. Allen et al. [50] and Hutter et al. [40] tried to conclude cell lineages or origins from transcription profiles of LCH cells. Cultured monocytes dramatically change their characters by adding factors such as a conditioned medium of an established LCH lesion cell line [56]. Very recently high-throughput single cell transcriptomics revealed different temporal heterogeneity profiles among identical mouse bone-marrow-derived DCs after same stimulation [57]. So it is not easy to conclude LCH cell lineages under the cytokine storm [16] using transcription profiles. So we use the terms mutant monocytes and mutant precursor LCs as precursor LCH cells in this review as shown in Figure 3.
Interleukin-1 autocrine/paracrine loop in LCH cells
The discovery of the B-Rapidly Accelerated Fibrosarcoma gene (BRAF) mutation in 2010 [20] gave new insights into LCH pathogenesis (Figures 3, 4, 5, 6, 7, and 8) and led to the hypothesis that the lesional IL-1 autocrine/paracrine loop [58,59] plays a major role in LCH pathogenesis as shown in Figures 3, 4, and 5 (IL-1 loop model).

Proposed relationship between MCPyV and mutated precursor LCs in LCH-risk organ (RO) (−). Mutated LCH precursor cells (mutant LC precursors) in peripheral tissues recognize MCPyV and induce LCH-RO (−). LCH lesion is consisted by accumulation and prolonged survival of LCH cells with T cells, eosinophils, macrophages, etc. In LCH lesions the prominent sources of cytokine storm are T cells and LCH cells. LCH cells produce pro-inflammatory cytokines such as IL-1, anti-inflammatory cytokines such as IL-10, and growth factors. After accumulation and prolonged survival of LCH cells, LCH lesions diminish and spontaneous healing may be observed.

Proposed relationship between MCPyV and mutated monocytes in LCH-RO (+). Mutated precursor LCH cells (mutant monocytes) in blood vessels recognize MCPyV and induce LCH-RO (+). LCH lesion is consisted by accumulation and prolonged survival of LCH cells with T cells, eosinophils, macrophages, etc., which is not different from that of LCH-RO (−). In LCH lesions the prominent sources of cytokine storm are T cells and LCH cells. LCH cells produce pro-inflammatory cytokines such as IL-1, anti-inflammatory cytokines such as IL-10, and growth factors. After accumulation and prolonged survival of LCH cells, LCH lesions diminish and spontaneous healing may be observed. But circulating monocytes with mutation triggered by MCPyV caused disseminated LCH lesions including RO.

Potential consequences of mutant BRAF V600E in LCH. In mutant monocytes or mutant LC precursors, the constitutively active BRAF V600E mutant protein is predicted to bypass the requirement for mitogen-induced activation of RAF by RAS. The identification of activating BRAF mutations supports the hypothesis that LCH is a neoplastic process (oncogenic potential). However, phosphorylated ERK is rapidly dephosphorylated by DUSP6, which is constitutively expressed in LCH cells (GSE16395). Other factors, such as accumulated gene mutations and an inflammatory trigger of the RAS/RAF/MEK/ERK signaling pathway, thus appear to be involved in LCH pathogenesis. DUSP6: dual specificity phosphatase 6.

Merkel cell polyomavirus is one candidate IL-1 trigger in LCH. Merkel cell polyomavirus (MCPyV) may be detected by Toll-like receptors (TLRs). MyD88 is a TLR adaptor protein that binds to pERK, maintaining ERK in an active, phosphorylated state for a longer period. Activated ERK phosphorylates numerous substrates related to the expression of soluble mediators such as IL-1β. Because of the low viral load of MCPyV-DNA in LCH tissue, MCPyV does not seem to play an oncogenic role in LCH pathogenesis. MCPyV is regarded as a potential trigger of IL-1β production. Although MyD88 usually allows the activation of NF-κB, MCPyV might interfere with NF-κB activation by targeting NF-κB essential modulator (NEMO). IL-1β is synthesized as an inactive pro-form (IL-1β precursor) that accumulates in the cytosol. Cleavage of IL-1β precursor into active form requires the activation of inflammasomes.

Proposed IL-1-loop model in LCH as a reactive disorder triggered by MCPyV. MyD88 is an adaptor of IL-1R and binds to pERK, maintaining ERK in an active, phosphorylated state. MyD88 also allows the activation of NF-κB, leading to the activation of further inflammatory and mitogenic signals. Induction of this IL-1β autocrine loop after MCPyV infection may lead to enhanced cell activation, proliferation, and eventually, transformation of LCH. In absence of MCPyV infection, the IL-1β paracrine loop also leads to enhanced cell activation, proliferation, and eventually, accumulation and cell survival of LCH cells. The clinical course of LCH may also be influenced by anti-inflammatory cytokines produced by T-cells under different conditions, including innate immunity alone and actuated acquired immunity against MCPyV.
The antigen-presentation capabilities of LCs are revealed only after IL-1β– and TNF-α–induced migration toward skin-draining lymph nodes [60,61]. LCH cells produce high levels of multiple cytokines, including IL-1 [62,63]. IL-1β is the first cytokine secreted in response to topical allergens via the inflammasome [64]; IL-1β mRNA can be detected in LCs as early as 15 min after exposure to sensitizers [65]. Although IL-1β is not produced under normal conditions, it is easily induced by slight stimulation, as shown by studies at the mRNA level [50]. Purification of LCs (Figure 1) from 4% of the entire cell population in the epidermis [66] to 97.3% [50] using anti-CD207 antibody was performed after incubation in RPMI 1640 with dispase II at 4°C for 8 h and 0.25% trypsin-EDTA for 15 min. Transient Receptor Potential (TRP) channels [67,68] are sensitive to temperature [69,70] and induce inflammasome activation [71]. In addition, the CD1a molecule is sensitive to trypsin [72]. Thus, this purification can induce LCs to produce IL-1β by comparing mean raw signals of IL-1β mRNA (log2) as follows: 8.8698 (LCs, n = 12), 9.379 (LCH cells of SS-LCH, n = 8), and 10.8729 (LCH cells of MS-LCH, n = 5) by re-analyses of GSE16395 [50] using Subio platform (http://www.subio.jp/products/platform) [39] (Figure 9). IL-1 stimulates MyD88, which activates nuclear factor-κB (NF-κB), leading to the subsequent activation of further inflammatory and mitogenic signals [73].

Line Graph of IL1B mRNA data from GSE16395. We compared GSE16395 mRNA array data between Langerhans cells (LCs) and LCH cells using the Subio platform. Each line represents a measured value. The red line represents IL1B mRNA expression, which is lower in both LCs and LCH cells according to the intensity of processed signals. However, using raw data signals, IL1B mRNA expression is high in both LCs and LCH cells. This phenomenon indicates that even slight stimuli, such as low temperature or tissue treatment using trypsin for LC isolation from the epidermis, can trigger IL-1β production. As previously reported, IL-1β expression was indicated. In addition IL-1β expression was higher in MS-LCH than SS-LCH.
Based on the serum and saliva levels of IL-1 [74,75], we advocate that the lesional IL-1 autocrine/paracrine loop [58,59] plays an important role in LCH pathogenesis, as shown in Figures 3, 4, and 5 (IL-1 loop model). This combination of MyD88-dependent signals may lead to enhanced cell activation, proliferation, and eventually, accumulation and prolonged cell survival [58,73] of LCH lesions.
Cigarette smoke and IL-1
Pulmonary LCH predominantly affects young adults and occurs almost exclusively in smokers [76-79]. Studies of cigarette smoke-induced accumulation of lung DCs in mice indicate an IL-1–dependent phenomenon [80]. In addition, cigarette smoke-induced pulmonary inflammation is dependent on Toll-like receptor (TLR) 4/MyD88 and IL-1R1/MyD88 signaling [81].
Merkel cell polyomavirus is a candidate for triggering the IL-1 loop involved in LCH pathogenesis
Patients with LCH often have dermal disorders such as seborrheic dermatitis [7] concomitant to LCH lesions [82], preceding LCH lesions [83-85], or following LCH lesions [86]. Perianal lesions [87,88] or a lesion on the soles [89] were also reported. Stein et al. [90] reported that children who present with LCH from birth to 4 weeks of age are not diagnosed with LCH until an average of 3.5 months of age because the eruptions are nonspecific in nature [91]. We recently described the possibility of a causal relationship between LCH and dermotropic Merkel cell polyomavirus (MCPyV) [92], which was discovered as the major pathogenic agent in Merkel cell carcinoma of the skin in 2008 [93]. Our data indicate that MCPyV-DNA sequences are present in LCH tissues (12/13) excluding pulmonary LCH, with significant differences between LCH tissues and controls that included patients with dermatopathic lymphadenopathy (5/20; P = .0002) and reactive lymphoid hyperplasia (0/5; P = .0007) [92]. The numbers of MCPyV DNA sequences in all four LCH tissues from patients younger than 2 years indicated a significant difference from tissues of non-LCH dermal disease patients of the same age (0/11; P = .0007) [92]. Our data suggest that LCH is a reactive disorder with an underlying oncogenic potential. Thus, both LCH and pulmonary LCH harbor the BRAF V600E mutation [20,94] and appear related to stimuli such as viral infection [92,95,96] and cigarette smoking [97,98]. In addition, the removal of stimuli is reported to cause spontaneous healing of LCH [99,100].
Expression of the constitutively active BRAF V600E mutant in LCH cells is predicted to bypass the requirement for mitogen-induced activation of RAF by RAS (Figure 6) [20,101]. The identification of activating BRAF mutations supports the hypothesis that LCH is a process with oncogenic potential [20]. A mouse LCH model using a BRAF V600E construct under control of the CD11c promoter and a BRAF V600E construct under control of the langerin promoter indicates that the BRAF V600E is not only a marker but also an essential driver of LCH pathogenesis [102]. Moreover, phosphorylated extracellular signal-regulated kinase (ERK) (pERK) is rapidly dephosphorylated by dual specificity phosphatase 6 (DUSP6) [73,103], which is overexpressed in LCH cells [50]. However, BRAF V600E mutations are also detected in non-neoplastic disorders such as nevus cell nevus (Figure 1) [104] and hyperplastic polyps of the colon [105]. Thus, LCH pathogenesis requires both limited proliferation of precursor LCH cells harboring the BRAF V600E mutation and the accumulation of gene mutations or an inflammatory trigger that activates the RAS/RAF/mitogen-activated protein kinase kinase (MEK)/ERK signaling pathway [101].
We reported the presence of MCPyV-DNA in the peripheral blood cells of two out of three patients with LCH-RO (+) but not in the blood cells of 12 of 12 patients with LCH-RO (−) (P = .029) [92]. Berres et al. [102] reported that patients with LCH-RO (+) carried the BRAF V600E mutation in circulating CD11c + and CD14+ cellular fractions as well as in BM CD34+ hematopoietic cell progenitors, whereas the mutation was restricted to lesional LCH cells in patients with LCH-RO (−). These findings specifically observed in LCH-RO (+) suggest the LCH pathogenetic pathway shown in Figures 3, 4, and 5, though it needs further confirmation to conclude.
MCPyV interferes with DC function (Figures 2 and 3) to evade immune surveillance by targeting a NF-κB essential modulator (NEMO) [106] and down-regulating TLR9 [107]. Exposure to MCPyV as measured by serum antibodies against the viral capsid proteins appears to be widely prevalent among healthy subjects [108,109]. Inapparent existence of MCPyV is indicated on the skin and environmental surface [110,111]. Pancaldi et al. [112] indicated buffy coats of healthy adult blood donors, which were examined for MCPyV DNA tag sequences, showed a prevalence of 22%, with viral loads ranging from 10 to 100 molecules per 100 000 cells (0.0001 to 0.001 per cell). Mertz et al. [113] reported that CD14 + CD16− inflammatory monocytes are a reservoir for MCPyV, but CD14loCD16+ resident monocytes, lymphocytes, or granulocytes are not. Our data from micro-dissected LC in both dermatopathic lymphadenopathy [92] and LC sarcoma [114] suggest that monocytes, precursor LCs, or LCs are one of the reservoir cells for MCPyV (Figure 3). Members of the TLR/IL-1 receptor (IL-1R) superfamily play a fundamental role in the immune response [115]. Viral “pathogen-associated molecular patterns” are recognized by specific TLRs [116]. TLR agonists stimulate IL-1β production in DC [117], where TLR-triggered ERK activation play important roles [118]. IL-1α expression is induced by TLR-mediated NF-κB activation; such activation has been observed in some LCH cases [119,120], with/without the presence of IL-1β [121]. All TLRs except TLR3 use the common MyD88-dependent pathway [122]. MyD88 is one of the adaptor proteins that links TLR/IL-1R [123] and binds to pERK via its D-domain, thereby preventing pERK–DUSP6 interaction and maintaining ERK in an active, phosphorylated state for a longer period [73]. This MyD88-dependent signal may lead to enhanced cell activation, proliferation, and eventually, accumulation and prolonged survival [58,73] of a given LCH lesion (Figure 8).
IL-1 family
The IL-1 family includes seven ligands with agonist activity (IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ), three receptor antagonists (IL-1Ra, IL-36Ra, and IL-38), and an anti-inflammatory cytokine (IL-37). Members of the IL-1R family include six receptor chains that form four signaling receptor complexes, two decoy receptors (IL-1R2 and IL-18BP), and two negative regulators (TIR8 or SIGIRR, and IL-1RAcPb). A tight regulation via receptor antagonists, decoy receptors, and signaling inhibitors ensures a balance between amplification of innate immunity and uncontrolled inflammation [124].
Our group reported that the serum level of IL-1α was significantly higher in patients with LCH than in controls [125], which suggests that IL-1 endocrine loop also plays an important role in LCH, in addition to the lesional IL-1 autocrine/paracrine loop. Serum levels of IL-18 were reported as significantly higher in LCH-RO (+) than in LCH-RO (−) [125]. Coury et al. [74] reported that serum levels of IL-1β in patients with LCH were not high; however, Preliasco et al. [75] reported IL-1β was increased in the saliva of children with LCH. Rosso et al. [126] reported that serum levels of IL-1Ra were significantly higher in patients with LCH than in controls. A tight regulation of cytokines via receptor antagonists such as IL-1Ra ensures a balance between amplification of innate immunity and uncontrolled inflammation. This balance may be overwhelmed by the cytokine storm caused by amplification of the IL-1 loop in BRAF mutant cells detected in patients with LCH (Figures 4 and 5). Agents inducing both IL-1β and IL-1Ra are viruses, bacteria, yeasts, soluble microbial products, IL-1, and TNF [127].
IL-1-mediated inflammation is also proposed to contribute to the development and progression of some cancers [73], such as melanoma [128,129]. The oncogenic BRAF V600E mutation promotes stromal cell-mediated immunosuppression via induction of IL-1 in melanoma [130]. Incisional biopsy of melanoma of the head and neck, which has a significant statistical association in the development of metastasis as compared to excisional biopsy [131], might be related to IL-1 expression through TRP channel triggered by mechanical stimulation [132,133]. IL-18 expression is also related to cancer-induced immunosuppression [134]. The following gene expressions are induced by IL-1: cytokines (TNF, IL-2, IL-3, IL-6, IL-12, GM-CSF, TGFβ, etc.); cytokine receptors (for IL-2, for IL-3, for IL-5, for GM-CSF, for c-kit); proinflammatory mediators (cyclooxygenase, etc.); hepatic acute phase reactants, clotting factors (fibrinogen, plasminogen activator inhibitor, etc.); oncogenes (c-jun, c-abl, c-fms, c-myc, c-fos) [127].
The Src homology region 2 domain-containing phosphatase −1 (SHP-1)
The tyrosine phosphatase SHP-1 plays an important role in DCs [135]. We thus investigated SHP-1 levels in LCH and found significantly higher expression of SHP-1 in MS-LCH than that in SS-LCH [38]. From this report, it has been proposed that SHP-1 might promote IL-1R/TLR–activated production of type I interferon, which is one of innate antiviral cytokines [136], by inhibiting IL-1R associated kinase 1 [137].
IL-17A receptor (IL-17RA)
Ziegler-Heitbrock et al. [138] proposed three types of monocytes: classical CD14++CD16− monocytes; intermediate CD14++CD16+ monocytes; nonclassical CD14 + CD16++ monocytes. Mertz et al. [113] reported that CD14 + CD16− inflammatory monocytes are a reservoir for MCPyV; they noted that circulating monocytes caused disseminated psoriatic lesion, in which IL-1 may be a key inflammatory mediator [139-141], over a course of 7 years in one patient who subsequently developed Merkel cell carcinoma. They [113] reported MCPyV DNA in a psoriatic lesion, too. Psoriasis is a skin disorder in which T lymphocytes and DCs play a central role [142]. IL-1 is a key mediator of psoriasis [139]. Viral infection such as vesicular stomatitis virus, vaccinia virus, and a variety of influenza A viruses triggers rapid differentiation of human blood monocytes into DCs [143]. Monocytes as a reservoir for MCPyV might have a potential to differentiate into DCs. T helper 17 cells (Th17) and their signature cytokine IL-17 have a critical role in the pathogenesis of psoriatic disease [144,145]. TLR2-activated human LCs promote Th17 polarization via IL-1β, TGF-β, and IL-23 [146] through MyD88 [147]. These data may suggest MCPyV could promote IL-1β via TLR of LCs. Inhibition of ERK was observed to suppress IL-1 and IL-23 production by DCs [118]. IL-1α and IL-6, which also stimulate Th17, were reported as significantly higher in LCH tissues [3,62,148,149] and cultured cells from LCH lesion [150], than in controls (P < 0.05) [125]. We detected higher levels of IL-17RA expression in MS-LCH than in SS-LCH [39] and proposed an IL-17 endocrine model that could settle the IL-17A controversy and the IL-17A paradox [74,151,152] in LCH pathogenesis.
Innate and adaptive immunity against MCPyV
As shown in Figures 3, 4, and 5, we propose different MCPyV infection patterns between LCH-RO (+) and LCH-RO (−) [92]. These patterns differ with respect to the presence or absence, respectively, of cells carrying the mutated BRAF V600E in BM or among circulating fractions [102]. We have observed that IL-18 serum levels are higher in LCH-RO (+) than in LCH-RO (−) [125]. IL-18 induces LC migration via TNF-α and IL-1β molecules [153]. Seroepidemiological surveys in Cameroon report MCPyV seroprevalence rates in children 1 year old and younger as 60% (0–2 months, n = 34), 70% (3–4 months, n = 11), 35% (5–10 months, n = 49), and 20% (11–12 months, n = 23) [154]. In contrast, the MCPyV seroprevalence rates in LCH patients aged 1 month to 1 year were 0% (n = 6), although the seroprevalence rates among LCH patients ≥2 years of age are similar to that of other groups [108,109]. Primary infection by MCPyV without maternal immunoglobulin against MCPyV might play a significant pathogenic role in LCH among patients aged 1 month to 1 year, when MS-LCH or LCH-RO (+) often occurs [2,3]. Innate immune function between newborns and elderly is extremely different and large quantities of IL-6 and IL-23 after TLR stimulation by term newborns are indicated [155]. Thus, IL-6 amplifier activation may also influence the clinical course of LCH [156]. Kumar et al. [157] found that MCPyV-specific Th-cells secrete the Th2-like cytokine IL-13, the regulatory cytokine IL-10 (anti-inflammatory cytokine), and the Th1-like cytokine IFN-γ (a major antiviral cytokine). IL-13 induces IL-1 Ra [158] whose levels in sera of LCH patients are reported to be high [126]. IL-10 prevents LC maturation and thought to contribute to the maintenance of LCH cells in an immature stage of differentiation [34]. The levels of these molecules were significantly higher among MCPyV-seropositive subjects than among seronegative subjects [157]. Senechal et al. [12] pointed out that compared with controls, the expansion of regulatory T-cells, which are inhibitory in adaptive immunity, is observed in both LCH lesion and peripheral blood of patients with LCH. Immune conditions including the release of regulatory T-cells and anti-inflammatory cytokines might modify the balance between amplification of innate immunity against MCPyV and subsequent uncontrolled inflammation.
Both innate and acquired immunity are regulated by the coordination of many molecules, including osteopontin (OPN) [159,160]. Expression of the OPN protein, encoded by SPP1 [50], is closely related to IL-1 levels [161-164]. We have recently reported high serum levels of OPN in LCH-RO (+) [165].
RAS/RAF/MEK/ERK signaling pathway in LCH cells
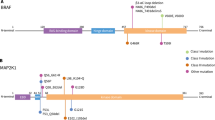
BRAF V600E mutations are present in 57% of all patients with LCH [20]. pERK was detected in LCH without the BRAF V600E mutation [20], indicating that other factors in the RAS/RAF/MEK/ERK signaling pathway may also be important in LCH pathogenesis. H-ras overexpression was first reported in LCH [166]. In cases of LCH without the BRAF V600E mutation, other mutations have been reported, including ARAF (one patient of two BRAF wild-type cases) [167] and MAP2K1 (MEK1) mutations [7/21 (33%) to 11/22 (50%) of BRAF wild-type cases] [168,169]. Very recently, Chacraborty et al. [169] reported an ERBB3 mutation.
Berres et al. [102] reported that hematopoietic progenitor cells are linked not only to LCH but also to juvenile xanthogranuloma or Erdheim–Chester disease (ECD), suggesting a common denominator for these conditions. Hervier et al. [170] also pointed out a relationship between LCH and ECD. NRAS mutations were detected in 3/17 ECD BRAF V600E wild-type patients, and PIK3CA mutations were detected in 7/55 patients, of whom 4 also had BRAF mutations [171]. As for cytokines, several studies supported a central role of the IL-1 network in ECD [172,173].
Other triggers of the IL-1 loop in LCH
Many factors including infectious agents and stress factors, inflammatory substances, and clotting factors are inducers of IL-1 [127]. In addition to MCPyV and cigarette smoking, Bacillus Calmette-Guerin (BCG) [174] and Epstein–Barr virus (EBV) infection [95,96] have been indicated as deeply involved in LCH pathogenesis. Induction of IL-1 by BCG [175-177] or EBV infection [178] was indicated.
Conclusions
We propose a new model for LCH pathogenesis in which the disease is a reactive disorder with underlying neoplastic potential. In other words, LCH is an inflammatory process that is prolonged by BRAF mutation. Spontaneous healing is more common in LCH and diminished activity of LCH cells might not be reversed by any more IL-1 at the later stage as shown in experiments using cultured LCH cells [179]. Triggers such as MCPyV stimulate cells with BRAF V600E mutations to produce IL-1 and a subsequent IL-1 autocrine/paracrine/endocrine loop cytokine storm. The state of acquired immunity against MCPyV may also influence the clinical course of LCH.
Together, these data indicate the importance of BRAF V600E mutations [180,181] as well as suggest that IL-1 could serve as a therapeutic target for LCH treatment, especially for prolonged inflammatory process supplied from circulating mutated monocytes in LCH-RO (+) [182]. IL-1R antagonists such as anakinra, already tried for ECD treatment [183,184], rilonacept, and canakinumab, with taking care of serious unwanted side-effects, might interrupt the out-of-control IL-1 loop, providing a therapeutic strategy for LCH treatment.
Abbreviations
- BM:
-
Bone marrow
- BRAF :
-
B-rapidlly accelerated fibrosarcoma gene
- DC:
-
Dendritic cell
- DUSP6:
-
Dual specificity phosphatase 6
- ECD:
-
Erdheim-Chester disease
- ERK:
-
Extracellular signal-regulated kinase
- GFR:
-
Growth factor receptor
- IL-1:
-
Interleukin 1
- IL-1R:
-
IL-1 receptor
- IL-1Ra:
-
IL-1 receptor antagonist
- IL-17RA:
-
IL-17A receptor
- LCs:
-
Langerhans cells
- LCH:
-
Langerhans cell histiocytosis
- LCH cells:
-
CD1a-positive abnormal LC-like cells
- LCH- RO (+):
-
LCH developed in at least one high-risk organ
- LCH-RO (−):
-
LCH without high-risk organ involvement
- MCPyV:
-
Merkel cell polyomavirus
- MEK:
-
mitogen–activated protein kinase kinase
- MMP:
-
Matrix metalloproteinase
- MS-LCH:
-
Multisystem LCH
- MyD:
-
Myeloid differentiation primary response
- NEMO:
-
Nuclear factor-κB essential modulator
- NF-κB:
-
Nuclear factor-κB
- OPN:
-
Osteopontin
- pERK:
-
phosphorylated ERK
- SS-LCH:
-
Single-system LCH
- Th:
-
T helper
- TNF-α:
-
Tumor necrosis factor-alpha
- TLR:
-
Toll-like receptor
- TRP:
-
Transient Receptor Potential
References
Chu T, D’Angio GJ, Favara B, Ladisch S, Nesbit M, Pritchard J. Histiocytosis syndromes in children. Lancet. 1987;1:208–9.
Hamre M, Hedberg J, Buckley J, Bhatia S, Finlay J, Meadows A, et al. Langerhans cell histiocytosis: an exploratory epidemiologic study of 177 cases. Med Pediatr Oncol. 1997;28:92–7.
Weitzman S, Egeler RM. Histiocytic disorders of children and adults: introduction to the problem, overview, historical perspective and epidemiology. In: Weitzman S, Egeler RM, editors. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press; 2005. p. 1–13.
Salotti JA, Nanduri V, Pearce MS, Parker L, Lynn R, Windebank KP. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child. 2009;94:376–80.
Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer. 2007;48:555–60.
Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51:71–5.
Donadieu J, Egeler RM, Pritchard J. Langerhans Cell Histiocytosis: A Clinical Update. In: Weitzman S, Egeler RM, editors. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press; 2005. p. 95–129.
Ronceray L, Potschger U, Janka G, Gadner H, Minkov M, German Society for Pediatric H, Oncology LCHSG. Pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on course and outcome. J Pediatr. 2012;161:129–33. e121-123.
Finn LS, Jaffe R. Langerhans’ cell granuloma confined to the bile duct. Pediatr Pathol Lab Med. 1997;17:461–8.
Kaplan KJ, Goodman ZD, Ishak KG. Liver involvement in Langerhans’ cell histiocytosis: a study of nine cases. Mod Pathol. 1999;12:370–8.
Delprat C, Arico M. Blood spotlight on Langerhans cell histiocytosis. Blood. 2014;124:867–72.
Senechal B, Elain G, Jeziorski E, Grondin V, de Patey-Mariaud Serre N, Jaubert F, et al. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med. 2007;4:e253.
Donadieu J. A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ Cell Histiocytosis Study Group. Arch Dis Child. 1996;75:17–24.
Bernard F, Thomas C, Bertrand Y, Munzer M, Landman Parker J, Ouache M, et al. Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur J Cancer. 2005;41:2682–9.
Bernstrand C, Sandstedt B, Ahstrom L, Henter JI. Long-term follow-up of Langerhans cell histiocytosis: 39 years’ experience at a single centre. Acta Paediatr. 2005;94:1073–84.
da Costa CE, Annels NE, Egeler RM. The Immunological Basis of Langerhans Cell Histiocytosis. In: Weitzman S, Egeler RM, editors. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press; 2005. p. 66–82.
Bhattacharjee P, Glusac EJ. Langerhans cell hyperplasia in scabies: a mimic of Langerhans cell histiocytosis. J Cutan Pathol. 2007;34:716–20.
Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, et al. Langerhans’-cell histiocytosis (histiocytosis X)–a clonal proliferative disease. N Engl J Med. 1994;331:154–60.
Yu RC, Chu C, Buluwela L, Chu AC. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet. 1994;343:767–8.
Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919–23.
Iwafuchi M, Watanabe H, Shiratsuka M. Primary benign histiocytosis X of the stomach. A report of a case showing spontaneous remission after 5 1/2 years. Am J Surg Pathol. 1990;14:489–96.
Corbeel L, Eggermont E, Desmyter J, Surmont I, De Vos R, De Wolf-Peeters C, et al. Spontaneous healing of Langerhans cell histiocytosis (histiocytosis X). Eur J Pediatr. 1988;148:32–3.
Bhatia S, Nesbit Jr ME, Egeler RM, Buckley JD, Mertens A, Robison LL. Epidemiologic study of Langerhans cell histiocytosis in children. J Pediatr. 1997;130:774–84.
Langerhans P. Uber die Nerven der menslichen Haut. Archive der Pathologischen Anatomie. 1868;44:325–37.
Coppes-Zantinga A, Egeler RM. The Langerhans cell histiocytosis X files revealed. Br J Haematol. 2002;116:3–9.
Holubar K, Kopera D. What is an eponym? Exemplified with remarks on Theodor Langhans, Friedrich Merkel, and Paul Langerhans, three contemporary 19th-century pathologists. J Invest Dermatol. 1994;103:257.
Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52.
Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–62.
Aiba S, Katz SI. Phenotypic and functional characteristics of in vivo-activated Langerhans cells. J Immunol. 1990;145:2791–6.
Fukunaga A, Nagai H, Yu X, Oniki S, Okazawa H, Motegi S, et al. Src homology 2 domain-containing protein tyrosine phosphatase substrate 1 regulates the induction of Langerhans cell maturation. Eur J Immunol. 2006;36:3216–26.
Aiba S. How are epidermal Langerhans cells activated in the initiation phase of allergic contact dermatitis? AATEX. 2005;11:49–58.
Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117:515–26.
Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30.
Geissmann F, Lepelletier Y, Fraitag S, Valladeau J, Bodemer C, Debre M, et al. Differentiation of Langerhans cells in Langerhans cell histiocytosis. Blood. 2001;97:1241–8.
Geissmann F, Emile JF, Andry P, Thomas C, Fraitag S, De Prost Y, et al. Lack of expression of E-cadherin is associated with dissemination of Langerhans’ cell histiocytosis and poor outcome. J Pathol. 1997;181:301–4.
Seo JJ, Cho T, Kim SY, Nassour I, Kim HJ, Lim YJ, et al. Prognostic significance of gelsolin and MMP12 in Langerhans cell histiocytosis. Korean J Hematol. 2012;47:267–72.
Battistella M, Fraitag S, Teillac DH, Brousse N, de Prost Y, Bodemer C. Neonatal and early infantile cutaneous langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149–56.
Murakami I, Oka T, Kuwamoto S, Kato M, Hayashi K, Gogusev J, et al. Tyrosine phosphatase SHP-1 is expressed higher in multisystem than in single-system Langerhans cell histiocytosis by immunohistochemistry. Virchows Arch. 2011;459:227–34.
Murakami I, Morimoto A, Oka T, Kuwamoto S, Kato M, Horie Y, et al. IL-17A receptor expression differs between subclasses of Langerhans cell histiocytosis, which might settle the IL-17A controversy. Virchows Arch. 2013;462:219–28.
Hutter C, Kauer M, Simonitsch-Klupp I, Jug G, Schwentner R, Leitner J, et al. Notch is active in Langerhans cell histiocytosis and confers pathognomonic features on dendritic cells. Blood. 2012;120:5199–208.
Wang B, Amerio P, Sauder DN. Role of cytokines in epidermal Langerhans cell migration. J Leukoc Biol. 1999;66:33–9.
Cumberbatch M, Dearman RJ, Kimber I. Interleukin 1 beta and the stimulation of Langerhans cell migration: comparisons with tumour necrosis factor alpha. Arch Dermatol Res. 1997;289:277–84.
Cumberbatch M, Bhushan M, Dearman RJ, Kimber I, Griffiths CE. IL-1beta-induced Langerhans’ cell migration and TNF-alpha production in human skin: regulation by lactoferrin. Clin Exp Immunol. 2003;132:352–9.
Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Ait-Yahia S, et al. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J Exp Med. 1998;188:373–86.
Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, Williams LT. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc Natl Acad Sci U S A. 1998;95:258–63.
Fleming MD, Pinkus JL, Fournier MV, Alexander SW, Tam C, Loda M, et al. Coincident expression of the chemokine receptors CCR6 and CCR7 by pathologic Langerhans cells in Langerhans cell histiocytosis. Blood. 2003;101:2473–5.
Polak ME, Thirdborough SM, Ung CY, Elliott T, Healy E, Freeman TC, et al. Distinct molecular signature of human skin Langerhans cells denotes critical differences in cutaneous dendritic cell immune regulation. J Invest Dermatol. 2014;134:695–703.
Kobayashi Y. Langerhans’ cells produce type IV collagenase (MMP-9) following epicutaneous stimulation with haptens. Immunology. 1997;90:496–501.
Ratzinger G, Stoitzner P, Ebner S, Lutz MB, Layton GT, Rainer C, et al. Matrix metalloproteinases 9 and 2 are necessary for the migration of Langerhans cells and dermal dendritic cells from human and murine skin. J Immunol. 2002;168:4361–71.
Allen CE, Li L, Peters TL, Leung HC, Yu A, Man TK, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010;184:4557–67.
Valladeau J, Saeland S. Cutaneous dendritic cells. Semin Immunol. 2005;17:273–83.
Valladeau J, Dezutter-Dambuyant C, Saeland S. Langerin/CD207 sheds light on formation of birbeck granules and their possible function in Langerhans cells. Immunol Res. 2003;28:93–107.
Feldman AL, Berthold F, Arceci RJ, Abramowsky C, Shehata BM, Mann KP, et al. Clonal relationship between precursor T-lymphoblastic leukaemia/lymphoma and Langerhans-cell histiocytosis. Lancet Oncol. 2005;6:435–7.
Magni M, Di Nicola M, Carlo-Stella C, Matteucci P, Lavazza C, Grisanti S, et al. Identical rearrangement of immunoglobulin heavy chain gene in neoplastic Langerhans cells and B-lymphocytes: evidence for a common precursor. Leuk Res. 2002;26:1131–3.
Murakami I, Takata K, Matsushita M, Nonaka D, Iwasaki T, Kuwamoto S, et al. Immunoglobulin expressions Are only associated with MCPyV-positive Merkel cell carcinomas but Not with MCPyV-negative ones: comparison of prognosis. Am J Surg Pathol. 2014;38:1627–35.
Murakami I, Gogusev J, Jaubert F, Matsushita M, Hayashi K, Miura I, et al. Establishment of a Langerhans cell histiocytosis lesion cell line with dermal dendritic cell characteristics. Oncol Rep. 2015;33:171–8.
Shalek AK, Satija R, Shuga J, Trombetta JJ, Gennert D, Lu D, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510:363–9.
Vale T, Ngo TT, White MA, Lipsky PE. Raf-induced transformation requires an interleukin 1 autocrine loop. Cancer Res. 2001;61:602–7.
Dinarello CA, Ikejima T, Warner SJ, Orencole SF, Lonnemann G, Cannon JG, et al. Interleukin 1 induces interleukin 1. I. Induction of circulating interleukin 1 in rabbits in vivo and in human mononuclear cells in vitro. J Immunol. 1987;139:1902–10.
Cumberbatch M, Dearman RJ, Kimber I. Langerhans cells require signals from both tumour necrosis factor-alpha and interleukin-1 beta for migration. Immunology. 1997;92:388–95.
Jakob T, Ring J, Udey MC. Multistep navigation of Langerhans/dendritic cells in and out of the skin. J Allergy Clin Immunol. 2001;108:688–96.
Egeler RM, Favara BE, van Meurs M, Laman JD, Claassen E. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195–201.
Sauder DN, Dinarello CA, Morhenn VB. Langerhans cell production of interleukin-1. J Invest Dermatol. 1984;82:605–7.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26.
Enk AH, Angeloni VL, Udey MC, Katz SI. An essential role for Langerhans cell-derived IL-1 beta in the initiation of primary immune responses in skin. J Immunol. 1993;150:3698–704.
Ackerman AB, Chongchitnant N, Sanchez J, Guo Y, Bennin B, Reichel M, et al. Embriologic, histologic, and anatomic aspects. In: Histologic diagnosis of inflammatory skin diseases. secondth ed. Baltimore: Williams and Wilkins; 1997.
Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417.
Szollosi AG, Olah A, Toth IB, Papp F, Czifra G, Panyi G, et al. Transient receptor potential vanilloid-2 mediates the effects of transient heat shock on endocytosis of human monocyte-derived dendritic cells. FEBS Lett. 2013;587:1440–5.
Vriens J, Nilius B, Voets T. Peripheral thermosensation in mammals. Nat Rev Neurosci. 2014;15:573–89.
Caterina MJ. Transient receptor potential ion channels as participants in thermosensation and thermoregulation. Am J Physiol Regul Integr Comp Physiol. 2007;292:R64–76.
Gross O, Yazdi AS, Thomas CJ, Masin M, Heinz LX, Guarda G, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400.
Dezutter-Dambuyant C, Schmitt D, Staquet MJ, Boumsell L, Thivolet J. Cleavage of Langerhans cell surface CD1a molecule by trypsin. Res Immunol. 1989;140:377–90.
Coste I, Le Corf K, Kfoury A, Hmitou I, Druillennec S, Hainaut P, et al. Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J Clin Invest. 2010;120:3663–7.
Coury F, Annels N, Rivollier A, Olsson S, Santoro A, Speziani C, et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med. 2008;14:81–7.
Preliasco VF, Benchuya C, Pavan V, de la Cal C, Ganzinelli S, Sterin-Borda L. IL-1 beta and PGE2 levels are increased in the saliva of children with Langerhans cell histiocytosis. J Oral Pathol Med. 2008;37:522–7.
Suri HS, Yi ES, Nowakowski GS, Vassallo R. Pulmonary langerhans cell histiocytosis. Orphanet J Rare Dis. 2012;7:16.
Vassallo R, Ryu JH, Schroeder DR, Decker PA, Limper AH. Clinical outcomes of pulmonary Langerhans’-cell histiocytosis in adults. N Engl J Med. 2002;346:484–90.
Arico M, Girschikofsky M, Genereau T, Klersy C, McClain K, Grois N, et al. Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003;39:2341–8.
Tazi A, Soler P, Hance AJ. Adult pulmonary Langerhans’ cell histiocytosis. Thorax. 2000;55:405–16.
Botelho FM, Nikota JK, Bauer CM, Morissette MC, Iwakura Y, Kolbeck R, et al. Cigarette smoke-induced accumulation of lung dendritic cells is interleukin-1alpha-dependent in mice. Respir Res. 2012;13:81.
Doz E, Noulin N, Boichot E, Guenon I, Fick L, Le Bert M, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol. 2008;180:1169–78.
Punia RS, Bagai M, Mohan H, Thami GP. Langerhans cell histiocytosis of skin: a clinicopathologic analysis of five cases. Indian J Dermatol Venereol Leprol. 2006;72:211–4.
Erdem AP, Kasimoglu Y, Sepet E, Gencay K, Sahin S, Dervisoglu S. Oral manifestations may be the first sign of Langerhans cell histiocytosis. Oral Health Prev Dent. 2013;11:57–9.
Mehta B, Venkatramani R. Images in clinical medicine. Langerhans’-cell histiocytosis. N Engl J Med. 2014;371:1050.
Lai JY, Tang MM, Priya G, Rajasuriar JS, Suganthi T. Langerhans Cell Histiocytosis in an Adult - a rare, life-threatening and not to be missed. Med J Malaysia. 2014;69:95–7.
Wada R, Yagihashi S, Konta R, Ueda T, Izumiyama T. Gastric polyposis caused by multifocal histiocytosis X. Gut. 1992;33:994–6.
Mittal T, Davis MD, Lundell RB. Perianal Langerhans cell histiocytosis relieved by surgical excision. Br J Dermatol. 2009;160:213–5.
Sabri M, Davie J, Orlando S, Di Lorenzo C, Ranganathan S. Gastrointestinal presentation of Langerhans cell histiocytosis in a child with perianal skin tags: a case report. J Pediatr Gastroenterol Nutr. 2004;39:564–6.
Guo W, Zhong LS, Wei ZP, Wu Q, Chen H. Case of Langerhans cell histiocytosis on soles. J Dermatol. 2014;41:946–7.
Stein SL, Paller AS, Haut PR, Mancini AJ. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med. 2001;155:778–83.
Krafchik B, Pope E, Walsh SRA. Histiocytosis of the Skin in Children and Adults. In: Weitzman S, Egeler RM, editors. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge University Press; 2005. p. 13–153.
Murakami I, Matsushita M, Iwasaki T, Kuwamoto S, Kato M, Horie Y, et al. Merkel cell polyomavirus DNA sequences in peripheral blood and tissues from patients with Langerhans cell histiocytosis. Hum Pathol. 2014;45:119–26.
Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–100.
Yousem SA, Dacic S, Nikiforov YE, Nikiforova M. Pulmonary Langerhans cell histiocytosis: profiling of multifocal tumors using next-generation sequencing identifies concordant occurrence of BRAF V600E mutations. Chest. 2013;143:1679–84.
Sakata N, Toguchi N, Kimura M, Nakayama M, Kawa K, Takemura T. Development of Langerhans cell histiocytosis associated with chronic active Epstein-Barr virus infection. Pediatr Blood Cancer. 2008;50:924–7.
Chen CJ, Ho TY, Lu JJ, Sheu LF, Lee SY, Tien CH, et al. Identical twin brothers concordant for Langerhans’ cell histiocytosis and discordant for Epstein-Barr virus-associated haemophagocytic syndrome. Eur J Pediatr. 2004;163:536–9.
Yousem SA, Colby TV, Chen YY, Chen WG, Weiss LM. Pulmonary Langerhans’ cell histiocytosis: molecular analysis of clonality. Am J Surg Pathol. 2001;25:630–6.
Tazi A, Hiltermann JN, Vassallo R. Adult Lung Histiocytosis. In: Weitzman S, Egeler RM, editors. Histiocytic Disorders of Children and Adults. Cambridge: Cambridge university press; 2005. p. 187–207.
Mogulkoc N, Veral A, Bishop PW, Bayindir U, Pickering CA, Egan JJ. Pulmonary Langerhans’ cell histiocytosis: radiologic resolution following smoking cessation. Chest. 1999;115:1452–5.
Von Essen S, West W, Sitorius M, Rennard SI. Complete resolution of roentgenographic changes in a patient with pulmonary histiocytosis X. Chest. 1990;98:765–7.
Nichols KE, Arceci RJ. BRAF, a piece of the LCH puzzle. Blood. 2010;116:1825–7.
Berres ML, Lim KP, Peters T, Price J, Takizawa H, Salmon H, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014;211:669–83.
Muda M, Boschert U, Dickinson R, Martinou JC, Martinou I, Camps M, et al. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J Biol Chem. 1996;271:4319–26.
Yeh I, von Deimling A, Bastian BC. Clonal BRAF mutations in melanocytic nevi and initiating role of BRAF in melanocytic neoplasia. J Natl Cancer Inst. 2013;105:917–9.
Chan TL, Zhao W, Leung SY, Yuen ST, Cancer Genome P. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res. 2003;63:4878–81.
Griffiths DA, Abdul-Sada H, Knight LM, Jackson BR, Richards K, Prescott EL, et al. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J Virol. 2013;87:13853–67.
Shahzad N, Shuda M, Gheit T, Kwun HJ, Cornet I, Saidj D, et al. The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J Virol. 2013;87:13009–19.
Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363.
Tolstov YL, Pastrana DV, Feng H, Becker JC, Jenkins FJ, Moschos S, et al. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer. 2009;125:1250–6.
Foulongne V, Kluger N, Dereure O, Mercier G, Moles JP, Guillot B, et al. Merkel cell polyomavirus in cutaneous swabs. Emerg Infect Dis. 2010;16:685–7.
Foulongne V, Courgnaud V, Champeau W, Segondy M. Detection of Merkel cell polyomavirus on environmental surfaces. J Med Virol. 2011;83:1435–9.
Pancaldi C, Corazzari V, Maniero S, Mazzoni E, Comar M, Martini F, et al. Merkel cell polyomavirus DNA sequences in the buffy coats of healthy blood donors. Blood. 2011;117:7099–101.
Mertz KD, Junt T, Schmid M, Pfaltz M, Kempf W. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus. J Invest Dermatol. 2010;130:1146–51.
Murakami I, Matsushita M, Iwasaki T, Kuwamoto S, Kato M, Horie Y, et al. High viral load of Merkel cell polyomavirus DNA sequences in Langerhans cell sarcoma tissues. Infect Agent Cancer. 2014;9:15.
Loiarro M, Ruggiero V, Sette C. Targeting TLR/IL-1R signalling in human diseases. Mediators Inflamm. 2010;2010:674363.
Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14.
He Y, Franchi L, Nunez G. TLR agonists stimulate Nlrp3-dependent IL-1beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol. 2013;190:334–9.
Brereton CF, Sutton CE, Lalor SJ, Lavelle EC, Mills KH. Inhibition of ERK MAPK suppresses IL-23- and IL-1-driven IL-17 production and attenuates autoimmune disease. J Immunol. 2009;183:1715–23.
Brown RE. The NF-kappaB pathway and the successful application of anti-inflammatory and angiostatic therapy in Langerhans’ cell histiocytosis. Br J Haematol. 2005;130:147–8.
Brown RE. Brief communication: morphoproteomic analysis of osteolytic Langerhans cell histiocytosis with therapeutic implications. Ann Clin Lab Sci. 2005;35:131–6.
Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, et al. Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc Natl Acad Sci U S A. 2011;108:18055–60.
Akira S, Yamamoto M, Takeda K. Role of adapters in Toll-like receptor signalling. Biochem Soc Trans. 2003;31:637–42.
Janssens S, Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002;27:474–82.
Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–18.
Morimoto A, Nakamura S, Shioda Y, Imamura T, Oh Y, Imashuku S, et al. Comprehensive analysis of serum levels of cytokines/chemokines and growth factors in pediatric patients with Langerhans cell histiocytosis [abstract]. Pediatr Blood Cancer. 2011;56:696.
Rosso DA, Ripoli MF, Roy A, Diez RA, Zelazko ME, Braier JL. Serum levels of interleukin-1 receptor antagonist and tumor necrosis factor-alpha are elevated in children with Langerhans cell histiocytosis. J Pediatr Hematol Oncol. 2003;25:480–3.
Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147.
Qin Y, Ekmekcioglu S, Liu P, Duncan LM, Lizee G, Poindexter N, et al. Constitutive aberrant endogenous interleukin-1 facilitates inflammation and growth in human melanoma. Mol Cancer Res. 2011;9:1537–50.
Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86.
Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18:5329–40.
Austin JR, Byers RM, Brown WD, Wolf P. Influence of biopsy on the prognosis of cutaneous melanoma of the head and neck. Head Neck. 1996;18:107–17.
Chen J, Barritt GJ. Evidence that TRPC1 (transient receptor potential canonical 1) forms a Ca(2+)-permeable channel linked to the regulation of cell volume in liver cells obtained using small interfering RNA targeted against TRPC1. Biochem J. 2003;373:327–36.
Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol. 2005;7:179–85.
Terme M, Ullrich E, Aymeric L, Meinhardt K, Coudert JD, Desbois M, et al. Cancer-induced immunosuppression: IL-18-elicited immunoablative NK cells. Cancer Res. 2012;72:2757–67.
Ramachandran IR, Song W, Lapteva N, Seethammagari M, Slawin KM, Spencer DM, et al. The phosphatase SRC homology region 2 domain-containing phosphatase-1 is an intrinsic central regulator of dendritic cell function. J Immunol. 2011;186:3934–45.
Iwasaki A, Medzhitov R. Innate Responses to Viral Infections. In: Knipe DM, Howley PM, editors. Fields Virology. sixthth ed. Philadelphia: Lippincott Williams & Wilkins, a Wolter Kluwer business; 2013. p. 189–213.
An H, Hou J, Zhou J, Zhao W, Xu H, Zheng Y, et al. Phosphatase SHP-1 promotes TLR- and RIG-I-activated production of type I interferon by inhibiting the kinase IRAK1. Nat Immunol. 2008;9:542–50.
Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–80.
Mee JB, Cork MJ, di Giovine FS, Duff GW, Groves RW. Interleukin-1: a key inflammatory mediator in psoriasis? Cytokine. 2006;33:72–8.
Cooper KD, Hammerberg C, Baadsgaard O, Elder JT, Chan LS, Taylor RS, et al. Interleukin-1 in human skin: dysregulation in psoriasis. J Invest Dermatol. 1990;95:24S–6.
Schon M, Behmenburg C, Denzer D, Schon MP. Pathogenic function of IL-1 beta in psoriasiform skin lesions of flaky skin (fsn/fsn) mice. Clin Exp Immunol. 2001;123:505–10.
Farkas A, Kemeny L. Monocyte-derived interferon-alpha primed dendritic cells in the pathogenesis of psoriasis: new pieces in the puzzle. Int Immunopharmacol. 2012;13:215–8.
Hou W, Gibbs JS, Lu X, Brooke CB, Roy D, Modlin RL, et al. Viral infection triggers rapid differentiation of human blood monocytes into dendritic cells. Blood. 2012;119:3128–31.
Raychaudhuri SP. Role of IL-17 in psoriasis and psoriatic arthritis. Clin Rev Allergy Immunol. 2013;44:183–93.
Kirkham BW, Kavanaugh A, Reich K. Interleukin-17A: a unique pathway in immune-mediated diseases: psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology. 2014;141:133–42.
Aliahmadi E, Gramlich R, Grutzkau A, Hitzler M, Kruger M, Baumgrass R, et al. TLR2-activated human langerhans cells promote Th17 polarization via IL-1beta, TGF-beta and IL-23. Eur J Immunol. 2009;39:1221–30.
Chang J, Burkett PR, Borges CM, Kuchroo VK, Turka LA, Chang CH. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc Natl Acad Sci U S A. 2013;110:2270–5.
de Graaf JH, Tamminga RY, Dam-Meiring A, Kamps WA, Timens W. The presence of cytokines in Langerhans’ cell histiocytosis. J Pathol. 1996;180:400–6.
Foss HD, Herbst H, Araujo I, Hummel M, Berg E, Schmitt-Graff A, et al. Monokine expression in Langerhans’ cell histiocytosis and sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). J Pathol. 1996;179:60–5.
Arenzana-Seisdedos F, Barbey S, Virelizier JL, Kornprobst M, Nezelof C. Histiocytosis X. Purified (T6+) cells from bone granuloma produce interleukin 1 and prostaglandin E2 in culture. J Clin Invest. 1986;77:326–9.
Peters TL, McClain KL, Allen CE. Neither IL-17A mRNA nor IL-17A protein are detectable in Langerhans cell histiocytosis lesions. Mol Ther. 2011;19:1433–9.
Allen CE, McClain KL. Interleukin-17A is not expressed by CD207(+) cells in Langerhans cell histiocytosis lesions. Nat Med. 2009;15:483–4. author reply 484–485.
Cumberbatch M, Dearman RJ, Antonopoulos C, Groves RW, Kimber I. Interleukin (IL)-18 induces Langerhans cell migration by a tumour necrosis factor-alpha- and IL-1beta-dependent mechanism. Immunology. 2001;102:323–30.
Martel-Jantin C, Pedergnana V, Nicol JT, Leblond V, Tregouet DA, Tortevoye P, et al. Merkel cell polyomavirus infection occurs during early childhood and is transmitted between siblings. J Clin Virol. 2013;58:288–91.
Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate immune function by Toll-like receptors: distinct responses in newborns and the elderly. Immunity. 2012;37:771–83.
Murakami M, Hirano T. The pathological and physiological roles of IL-6 amplifier activation. Int J Biol Sci. 2012;8:1267–80.
Kumar A, Chen T, Pakkanen S, Kantele A, Soderlund-Venermo M, Hedman K, et al. T-helper cell-mediated proliferation and cytokine responses against recombinant Merkel cell polyomavirus-like particles. PLoS One. 2011;6:e25751.
Vannier E, de Waal Malefyt R, Salazar-Montes A, de Vries JE, Dinarello CA. Interleukin-13 (IL-13) induces IL-1 receptor antagonist gene expression and protein synthesis in peripheral blood mononuclear cells: inhibition by an IL-4 mutant protein. Blood. 1996;87:3307–15.
Wang KX, Denhardt DT. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008;19:333–45.
Lund SA, Giachelli CM, Scatena M. The role of osteopontin in inflammatory processes. J Cell Commun Signal. 2009;3:311–22.
Yu XQ, Fan JM, Nikolic-Paterson DJ, Yang N, Mu W, Pichler R, et al. IL-1 up-regulates osteopontin expression in experimental crescentic glomerulonephritis in the rat. Am J Pathol. 1999;154:833–41.
Jin CH, Miyaura C, Ishimi Y, Hong MH, Sato T, Abe E, et al. Interleukin 1 regulates the expression of osteopontin mRNA by osteoblasts. Mol Cell Endocrinol. 1990;74:221–8.
Serlin DM, Kuang PP, Subramanian M, O’Regan A, Li X, Berman JS, et al. Interleukin-1beta induces osteopontin expression in pulmonary fibroblasts. J Cell Biochem. 2006;97:519–29.
Naldini A, Leali D, Pucci A, Morena E, Carraro F, Nico B, et al. Cutting edge: IL-1beta mediates the proangiogenic activity of osteopontin-activated human monocytes. J Immunol. 2006;177:4267–70.
Oh Y, Morimoto A, Shioda Y, Imamura T, Kudo K. Imashuku S, for the Japan LCHSG. High serum osteopontin levels in pediatric patients with high risk Langerhans cell histiocytosis Cytokine. 2014;70:194–7.
Abdelatif OM, Chandler FW, Pantazis CG, McGuire BS. Enhanced expression of c-myc and H-ras oncogenes in Letterer-Siwe disease. A sequential study using colorimetric in situ hybridization. Arch Pathol Lab Med. 1990;114:1254–60.
Nelson DS, Quispel W, Badalian-Very G, van Halteren AG, van den Bos C, Bovee JV, et al. Somatic activating ARAF mutations in Langerhans cell histiocytosis. Blood. 2014;123:3152–5.
Brown NA, Furtado LV, Betz BL, Kiel MJ, Weigelin HC, Lim MS, et al. High prevalence of somatic MAP2K1 mutations in BRAF V600E negative Langerhans cell histiocytosis. Blood. 2014;124:1655–8.
Chakraborty R, Hampton OA, Shen X, Simko S, Shih A, et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014;124:3007–15.
Hervier B, Haroche J, Arnaud L, Charlotte F, Donadieu J, Neel A, et al. Association of both Langerhans cell histiocytosis and Erdheim-Chester disease linked to the BRAFV600E mutation. Blood. 2014;124:1119–26.
Emile JF, Diamond EL, Helias-Rodzewicz Z, Cohen-Aubart F, Charlotte F, Hyman DM, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood. 2014;124:3016–9.
Tran TA, Pariente D, Lecron JC, Delwail A, Taoufik Y, Meinzer U. Treatment of pediatric Erdheim-Chester disease with interleukin-1-targeting drugs. Arthritis Rheum. 2011;63:4031–2.
Aouba A, Georgin-Lavialle S, Pagnoux C, Martin Silva N, Renand A, Galateau-Salle F, et al. Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood. 2010;116:4070–6.
Numakura S, Morikawa T, Ushiku T, Toyoshima T, Fukayama M. Langerhans cell histiocytosis of the urinary bladder in a patient with bladder cancer previously treated with intravesical Bacillus Calmette-Guerin therapy. Pathol Res Pract. 2014;210:123–6.
De Boer EC, De Jong WH, Steerenberg PA, Aarden LA, Tetteroo E, De Groot ER, et al. Induction of urinary interleukin-1 (IL-1), IL-2, IL-6, and tumour necrosis factor during intravesical immunotherapy with bacillus Calmette-Guerin in superficial bladder cancer. Cancer Immunol Immunother. 1992;34:306–12.
Conti P, Reale M, Nicolai M, Barbacane RC, Placido FC, Iantorno R, et al. Bacillus Calmette-Guerin potentiates monocyte responses to lipopolysaccharide-induced tumor necrosis factor and interleukin-1, but not interleukin-6 in bladder cancer patients. Cancer Immunol Immunother. 1994;38:365–71.
Luo Y, Chen X, O’Donnell MA. Mycobacterium bovis bacillus Calmette-Guerin (BCG) induces human CC- and CXC-chemokines in vitro and in vivo. Clin Exp Immunol. 2007;147:370–8.
D’Addario M, Ahmad A, Xu JW, Menezes J. Epstein-Barr virus envelope glycoprotein gp350 induces NF-kappaB activation and IL-1beta synthesis in human monocytes-macrophages involving PKC and PI3-K. FASEB J. 1999;13:2203–13.
Yu RC, Alaibac M, Chu AC. Functional defect in cells involved in Langerhans cell histiocytosis. Arch Dermatol Res. 1995;287:627–31.
Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121:1495–500.
Charles J, Beani JC, Fiandrino G, Busser B. Major response to vemurafenib in patient with severe cutaneous Langerhans cell histiocytosis harboring BRAF V600E mutation. J Am Acad Dermatol. 2014;71:e97–9.
Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–32.
Munoz J, Janku F, Cohen PR, Kurzrock R. Erdheim-Chester disease: characteristics and management. Mayo Clin Proc. 2014;89:985–96.
Mazor RD, Manevich-Mazor M, Shoenfeld Y. Erdheim-Chester Disease: a comprehensive review of the literature. Orphanet J Rare Dis. 2013;8:137.
Acknowledgments
The authors would like to thank all patients, parents, control subjects, and physicians who participated in the studies related to this review. These works were partly supported by the Histiocytosis Association of America (HAA grant 2009); Grants-in-aid for Scientific Research (C) 23590426 and (C) 26460451 from the Japanese Ministry of Education, Science, Sports and Culture; and by 2010, 2011, and 2012 research grants from the Japan LCH Study Group.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
IM, KH, and JG wrote the manuscript and prepared all the figures. MM, TI, SK, MK, KN, JG, FJ, KT, and TO participated in experimental analyses. YH, TI, AM, and SI provided materials and clinical data from patients with LCH. KH, FJ, SI, and TY supervised the project. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Murakami, I., Matsushita, M., Iwasaki, T. et al. Interleukin-1 loop model for pathogenesis of Langerhans cell histiocytosis. Cell Commun Signal 13, 13 (2015). https://doi.org/10.1186/s12964-015-0092-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-015-0092-z