
Abstract
Background
Increased apolipoprotein (apo) B level (hyperapoB) is a strong predictor of cardiovascular disease (CVD), even in patients who achieve recommended LDL-Cholesterol (LDL-C) goals. ApoB level, an important correlate of metabolic syndrome (MetS), is influenced by several gene-environment interactions. Some of them are rare and can explain a large proportion of apoB variance, whereas others more common have variable effects. The aim of this study was to evaluate the association of interaction between smoking and common hyperapoB gene variants (PPARα-L162V, lipoprotein lipase loss-of function mutation, apo e4 allele or apo E2/2 genotype) with plasma apoB concentrations, according to the expression of MetS.
Methods
This study was performed among 1798 subjects. Smoking was defined as non/mild smokers vs. moderate-to-heavy smokers. ApoB levels were determined using nephelometry. Logistic regression models were used to document interactions between smoking habits and the presence of hyperapoB gene variants on the relative odds to exhibit increased plasma apoB concentrations.
Results
Around 29% of individuals with a low-risk lipid profile without MetS component had hyperapoB. Smoking and the presence of hyperapoB gene variants tended to be associated with higher plasma apoB levels even in presence of low-LDL-C. There was a significant interaction (P = 0.04) between the presence of ≥1 gene variants and smoking on the risk to exhibit hyperapoB among subjects with low risk profile in primary prevention.
Conclusions
Combination of life habits assessment and some common genes variants may detect a significant proportion of patients with increased apoB levels, and therefore a higher risk of CVD, who could have been initially perceived as low-risk.
Similar content being viewed by others


Background
Low-density lipoprotein-cholesterol (LDL-C) and non-high-density lipoprotein-cholesterol (non-HDL-C) are well-known risk factors of cardiovascular disease (CVD) and important targets for intervention. Although the interest given to increased plasma apolipoprotein (apo) B concentrations is more recent, it has long been proven to also be an efficient marker of CVD risk in various studies and is now starting to be used as such [1,2,3]. In fact, the predictive power of hyperapobetalipoproteinemia (hyperapoB) would remain significant even in individuals with low-to-normal LDL-C [4].
Plasma apoB concentration is affected by a huge number of genetic factors, but most of those having a large size effect, such as familial hypercholesterolemia (FH)-causing gene variants, are not very frequent [5, 6]. However, the effect of most genetic variants is much more modest. ApoE gene polymorphisms, the peroxisome proliferator-activated receptor (PPAR)α-L162V mutation, as well as loss-of-function (LoF) lipoprotein lipase (LPL) gene mutations, are examples of common variants modulating apoB-containing lipoprotein metabolism. PPARα-L162V mutation and several LoF LPL gene variants have been associated with higher apoB levels [7, 8]. The differential contribution of common apoE polymorphisms (APOE2, 3 and 4) to apoB-containing lipoprotein metabolism is also well documented. APOE4 is associated with higher apoB, LDL-C levels and cholesterol absorption/synthesis ratio than APOE3. APOE2, in the homozygous state, is associated with a reduced very-low-density lipoprotein (VLDL)-remnants clearance, an increased number of intermediate-density lipoprotein (IDL) particles (β-VLDL) and a higher concentration of IDL-apoB, increasing the risk of dysbetalipoproteinemia and CVD [9].
ApoB is closely related to the expression of the metabolic syndrome (MetS), a worldwide increasingly prevalent phenotype highly influenced by life habits [10, 11]. While the literature abounds with studies on associations between apoB levels and life habits such as diet, alcohol and physical activity, it is less the case with smoking. According to available results, effects of smoking on lipid metabolism appear more obviously related to the HDL and triglycerides (TG) metabolism [12,13,14,15]. Fewer results target its association with apoB and these are not all in agreement [16,17,18].
An increasing number of gene-smoking interaction studies and transcriptome analyses have been published so that there is now abundant evidence about the interaction between smoking and gene factors in several metabolic pathways [19,20,21]. However, there are very few results and numerous disagreements about gene x smoking interactions influence on the lipid-lipoprotein metabolism, especially regarding apoB [22, 23]. Thus, while the relative CVD risk associated with rare monogenic conditions such as FH is recognized, the risk of different combinations of common genetic variants and environmental factors is still quite less known.
The aim of this study was to evaluate the association of interactions between smoking and common gene variants affecting apoB catabolic pathways (PPARα-L162V, heterozygous loss-of function LPL mutation, apo e4 allele or apo E2/2 genotype) with plasma apoB concentrations, according to the expression of MetS.
Methods
Subjects and clinical data
This study comprised a sample of 1798 French Canadians subjects from the Saguenay–Lac-Saint-Jean region of Quebec (Canada). All subjects were screened at the Chicoutimi Hospital Lipid Clinic or ECOGENE-21 Clinical Research Center and agreed to participate in studies on genetic determinants of type 2 diabetes or coronary artery disease (CAD) combining genome wide scans and candidate gene strategies. Subjects were selected to be included in the present study based on the availability of data on targeted apoB-associated mutations/polymorphisms, plasma apoB concentrations and smoking habits. Subjects with familial chylomicronemia (complete LPL deficiency) and those taking drugs known to affect blood lipid levels were excluded. Subjects were classified according to the MetS diagnosis, as confirmed by the presence of ≥3/5 of the following components: waist circumference > 102 cm in men or > 88 cm in women; fasting plasma TG concentrations ≥1.7 mmol/L; HDL-C < 1.0 mmol/L in men or < 1.3 mmol/L in women; blood pressure ≥ 130 mmHg or ≥ 85 mmHg for systolic or diastolic blood pressure, respectively, or hypertensive treatment; fasting glucose > 5.6 mmol/L or drug treatment for elevated glucose [10]. The presence of CAD was documented using patient’s medical charts on the basis of clinical and electrocardiography (ECG) criteria of myocardial infarction or evidence of coronary stenosis of at least 50% in > 1 main coronary arteries following coronary angiography for the investigation of ischemic heart disease. Waist girth was determined according to the procedures of the Airlie conference [24]. Smoking habits were classified according to the number of cigarettes smoked daily (0 to 10 (non/mild smokers) vs. more than 10 (moderate-to-heavy smokers)). Subjects gave their informed consent to participate in this study and were assigned a code that systematically de-identifies all clinical data [25]. This study has received the approval of the Chicoutimi Hospital Ethics Committee, and was conducted in accordance with the Declaration of Helsinki.
Biochemical analysis
Blood samples were obtained after a 12-h overnight fast from the antecubital vein into vacutainer tubes containing EDTA. Cholesterol, TG and glucose levels were measured by enzymatic assays on a CX7Analyser (Beckman, Fullerton, CA, USA) [26]. Total cholesterol was determined in plasma and HDL after precipitation of VLDL and LDL (d > 1.006 g/ml) in the infranatant with heparin and manganese chloride (MnCl2). In this case, plasma LDL cholesterol levels were estimated using the Friedewald formula [27] unless TG level was higher than 4.5 mmol/L, in which case a direct measurement was used. ApoB levels were determined using nephelometry.
Genotyping
LoF LPL gene variants included those with a combined prevalence of at least 5% in the Eastern Quebec French Canadian Founder population (P207L, G188E) or reported as prevalent worldwide (D9N and N291S). The PPARα-L162V variant, the presence of P207L, G188E, D9N and N291S variants in the LPL gene and the apo E genotype were identified by a mismatch polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) based method, as previously described [28,29,30,31,32,33].
Statistical analysis
Categorical variables were compared using the Pearson χ2 statistic, whereas group differences for continuous variables were compared with the Student’s unpaired two-tailed t–test. Log10-transformed data and medians (interquartile ranges) were used for variables with a non-normal distribution. Multivariate logistic regression models were built to document the relation between increase in the number of MetS components and the proportion of subjects with apoB levels > 0.9 g/L as well as to calculate significance of interactions between smoking habits and the presence of hyperapoB genotypes on the relative odds (odds ratio (OR)) to exhibit plasma apoB concentrations > 0.9 mmol/L. Age and gender were included as covariates. All statistical analyses were performed with the SPSS package (20.0. Armonk, NY: IBM Corp).
Results
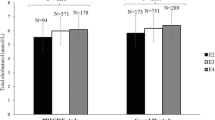
As shown in Table 1, subjects with MetS were older, had higher concentrations of total apoB, an increased frequency of CAD (P < 0.001) and were more frequently carriers of a loss-of-function LPL gene mutation (P = 0.001). In both group, almost 50% of subjects were not carrying any of the mutation (or polymorphism) known to be associated with increase in apoB concentration. The proportion of subjects with plasma apoB > 0.9 g/L significantly rises with the number of MetS components but only when LDL-C is < 3.5 mmol/L (P < 0.001). Increase in the number of MetS components is associated with an OR of 1.8 (P < 0.001) to exhibit plasma apoB > 0.9 mmol/L among subjects with LDL-C < 3.5 mmol/L, whereas the relation between both variables is non-significant in subjects with LDL-C ≥ 3.5 mmol/L (OR = 1.1; P = 0.58). In the LDL-C < 3.5 mmol/L group, more than one fourth (29%) of individuals without any MetS component had plasma apoB > 0.9 g/L (Fig. 1).

Proportion of subjects (%) with plasma apoB > 0.9 g/L, according to LDL-C and the number of metabolic syndrome components. Data were age-adjusted. NS = P > 0.1
As shown in Table 2, there is a significant interaction between the presence of ≥1 hyperapoB variants, in either the LPL, APOE, or PPARα genes, and smoking habits on the risk to exhibit apoB > 0.9 g/L, but only among subjects with a low risk profile, that is without MetS and with LDL-C < 3.5 mmol/L. Interestingly, this interaction remains significant even among individuals that never had CAD who are in primary prevention (Table 3).
Discussion
This study shows that a significant proportion of individuals have increased plasma apoB levels > 0.9 g/L even when LDL-C levels are not elevated and in absence of any criterion of the MetS. This observation is in accordance with previous results suggesting that not only can high apoB concentrations be observed in an important proportion of individuals with “optimal” LDL-C [34] but also that increases in cholesterol-depleted apoB particles can precede the onset of obesity. Hypertriglyceridemia and hyperglycemia (and then potentially the MetS) can therefore be an important marker of future CVD [35]. Current results are consistent with this statement. This study however goes a step further in the identification of potential markers of increased CVD risk, before the increase of apoB concentrations beyond high-risk levels. It also shows that common gene-smoking interactions are significantly associated with an increased risk of higher apoB concentrations among subjects with an apparently low-risk profile without history of CAD and who are in primary prevention.
The influence of smoking habits and of smoking x gene interactions on atherosclerotic processes, including inflammation, fat peroxidation, brown fat metabolism and coronary artery calcification has been relatively well documented in past years. Most studies get the same observation: smoking by itself is an important risk factor and, in addition, it significantly interacts with many gene variants by modulating the risk of CAD, CVD and stroke [19, 20]. Various studies have also suggested that smoking may influence apolipoproteins concentrations and lipoprotein particle sizes, although available results still remain limited and somewhat discordant [16, 17, 36]. Besides, there are fewer published results about gene x smoking interaction influence on lipid-lipoprotein metabolism, and there is even more disagreement between published observations [22, 23]. Interestingly, a recent large, multi-ancestry, genome-wide gene-smoking interaction study has reported 13 novel loci associated with lipid levels [37]. However, there are still apparent discrepancies between available results. Such a situation is not surprising considering the challenges associated with gene-environment interaction studies due to the difficulty to accurately control environment factors and to uniformly evaluate and characterize the studied disorders, especially for continuous traits such as lipid-lipoprotein phenotypes.
Study of subjects without apparent lipid-lipoprotein abnormalities or related disorders gave a particular clinical significance to the current results. They suggest that the assessment of life habits and the genotyping of some common gene variants may detect several patients with increased apoB measurements, and thus at a higher risk of CVD, that could have fallen of the radar with traditional guidelines. They also give more strength and solid arguments for preventive approaches, especially among people who do not believe themselves exposed to an increased CVD risk. This study included subjects whose average age was in the fifties and then who has been exposed to smoking for several years. Preventive strategies that could arise from such results could be more difficult to implement with subjects whose firmly fixed life habits may be more difficult to change. Besides, it has been shown that even a significant number of patients with proven coronary heart disease continue to smoke [38, 39]. However, results obtained from previous studies have shown that, even when young, smokers’ lipoproteins are altered as compared to non-smokers [40]. Although the present study was not conducted in young people, it could be hypothesize that similar results would be observed in younger subjects. It has been show that the cardiovascular risk associated with increased apoB levels would be higher among people under 50 [41]. Implementation of preventives strategies would therefore be of particular importance even among younger people.
The current results are in accordance with previous studies suggesting that smoking may alter the expression of genes influencing HDL metabolism or TG and LDL particle size [42]. Each of the genetic modulators included in this study, namely PPARα-L162V, heterozygous loss-of function LPL mutation, apo e4 allele or apo E2/2 genotype, is associated with a plasma apoB concentration increase, by acting on specific apoB-containing lipoproteins [7,8,9]. Various mechanisms could explain this interaction that could be partly due, for instance, to the unfavorable interaction between apo E4 and smoking, as well as to a reduced LPL activity previously observed among smokers [43,44,45]. Moreover, it has been shown that smoking is associated with an increased hepatic lipase activity [45, 46]. This increase, along with an elevation of TG-rich lipoprotein concentrations induced by the presence of the hyperapo B genotypes herein studied, could promote LDL-TG hydrolysis by hepatic lipase, which results in the generation of small-dense LDL particles and, then, in an increase of plasma apo B levels [47]. The epigenetic signature of smoking is also another interesting avenue to further investigate. In addition to the epigenetic effects of smoking on cancer physiopathology, there is increasing number of results showing significant associations between tobacco smoking and DNA methylation of genes associated with MetS components and associated risk [48].
The present study was conducted among a uniformly detailed phenotyped sample. This is one of its greatest strength. In addition, the French-Canadian population of the Saguenay–Lac-Saint-Jean region, from which the participants originate from, is another strength. This population is descended from a founder population that settled this region 300–400 years ago. This founder effect provides several benefits for studying the genomic determinants of complex traits. Genetic heterogeneity remains a problem in gene association studies that can be avoided by analyzing homogeneous populations that have a geographic stability and are most likely uniform in their environmental exposure [49].
One limitation of the current study is its modest sample size. It doesn’t allow the various subdivisions that would help to better document the associations observed, for instance a finer subdivision for the number of cigarettes smoked daily. Other life habits, including diet, which is clearly a central modulator in plasma lipid concentrations and can be related to smoking habits, was not available [50]. Finally, the cross-sectional design of the current study does not give any information about the potential causal pathway that may be implicated and the association with future CVD. Results should therefore be replicated in larger samples and diversified populations, with a further assessment of other life habits and, ideally, with the longitudinal follow-up of subjects to evaluate the hazard ratio of CVD.
Conclusion
ApoB levels, now recognized as an efficient marker of CVD risk, could be beyond clinical cut-off levels among low-risk subjects without apparent lipid-lipoprotein abnormalities. Obtained results are in accordance with the fact that common genetic combinations involving key players in the VLDL catabolic pathway and their interactions with smoking habits, are associated with an increased risk of elevated apoB. In conclusion, the current study shows that the combination of the assessment of life habits and some common genes variants may detect a significant proportion of patients with increased apoB levels, and therefore at a higher risk of CVD, who could have been perceived as low-risk initially.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Apo:
-
Apolipoprotein
- CAD:
-
Coronary artery disease
- CVD:
-
Cardiovascular disease
- FH:
-
Familial hypercholesterolemia
- HyperapoB:
-
Hyperapobetalipoproteinemia
- IDL:
-
Intermediate-density lipoprotein
- LDL-C:
-
Low-density lipoprotein-cholesterol
- LoFL:
-
Loss-of-function
- LPL:
-
Lipoprotein lipase
- MnCl2:
-
Heparin and manganese chloride
- Non-HDL-C:
-
Non-high-density lipoprotein-cholesterol
- OR:
-
Odds ratio
- PCR-RFLP:
-
Polymerase chain reaction-restriction fragment length polymorphism
- PPAR:
-
Peroxisome proliferator-activated receptor
- TG:
-
Triglycerides
- VLDL:
-
Very-low-density lipoprotein
References
Stock JK. New joint consensus initiative on quantifying atherogenic lipoproteins. Atherosclerosis. 2018;279:97–9.
Anderson TJ, Grégoire J, Pearson GJ, Barry AR, Couture P, Dawes M, et al. Canadian cardiovascular society guidelines for the Management of Dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol. 2016;32:1263–82.
Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, et al. 2016 ESC/EAS guidelines for the Management of Dyslipidaemias. Eur Heart J. 2016;37:2999–3058.
Sniderman AD, Islam S, Yusuf S, McQueen MJ. Discordance analysis of apolipoprotein B and non-high density lipoprotein cholesterol as markers of cardiovascular risk in the INTERHEART study. Atherosclerosis. 2012;225:444–9.
Santos RD, Maranhao RC. What is new in familial hypercholesterolemia? Curr Opin Lipidol. 2014;25:183–8.
Reiner Ž. Management of patients with familial hypercholesterolaemia. Nat Rev Cardiol. 2015;12:565–75.
Robitaille J, Brouillette C, Houde A, Lemieux S, Pérusse L, Tchernof A, et al. Association between the PPARalpha-L162V polymorphism and components of the metabolic syndrome. J Hum Genet. 2004;49:482–9.
Ooi EM, Russell BS, Olson E, Sun SZ, Diffenderfer MR, Lichtenstein AH, et al. Apolipoprotein B-100-containing lipoprotein metabolism in subjects with lipoprotein lipase gene mutations. Arterioscler Thromb Vasc Biol. 2012;32:459–66.
Ooi EM, Janus ED, Grant SJ, Sinclair LM, R Barrett PH. Effect of apolipoprotein E genotype on apolipoprotein B-100 metabolism in normolipidemic and hyperlipidemic subjects. J Lipid Res. 2010;51:2413–21.
Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. International Diabetes Federation Task Force on Epidemiology and Prevention; Hational Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; International Association for the Study of Obesity. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5.
Sattar N, Williams K, Sniderman AD, D’Agostino R Jr, Haffner SM. Comparison of the associations of apolipoprotein B and non-high-density lipoprotein cholesterol with other cardiovascular risk factors in patients with the metabolic syndrome in the insulin resistance atherosclerosis study. Circulation. 2004;110:2687–93.
Maeda K, Noguchi Y, Fukui T. The effects of cessation from cigarette smoking on the lipid and lipoprotein profiles: a meta-analysis. Prev Med. 2003;37:283–90.
Bermudez V, Olivar LC, Torres W, Navarro C, Gonzalez R, Espinoza C, et al. Cigarette smoking and metabolic syndrome components: a cross-sectional study from Maracaibo City, Venezuela. Version 3. F1000Res. 2019;7:565.
Jain RB, Ducatman A. Associations between smoking and lipid/lipoprotein concentrations among US adults aged ≥20 years. J Circ Biomark. 2018. https://doi.org/10.1177/1849454418779310.
Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Borén J, Catapano AL, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–61.
Frondelius K, Borg M, Ericson U, Borné Y, Melander O, Sonestedt E. Lifestyle and dietary determinants of serum Apolipoprotein A1 and Apolipoprotein B concentrations: cross-sectional analyses within a Swedish cohort of 24,984 individuals. Nutrients. 2017;9:E211.
Jain RB. Impact of smoking on the observed levels of apolipoprotein B: data from NHANES 2007-2012. Environ Toxicol Pharmacol. 2017;53:227–33.
Meenakshisundaram R, Rajendiran C, Thirumalaikolundusubramanian P. Lipid and lipoprotein profiles among middle aged male smokers: a study from southern India. Tob Induc Dis. 2010;8:11.
Merhi M, Demirdjian S, Hariri E, Sabbah N, Youhanna S, Ghassibe-Sabbagh M, et al. Impact of inflammation, gene variants, and cigarette smoking on coronary artery disease risk. Inflamm Res. 2015;64:415–22.
Polfus LM, Smith JA, Shimmin LC, Bielak LF, Morrison AC, Kardia SL, et al. Genome-wide association study of gene by smoking interactions in coronary artery calcification. PLoS One. 2013;8:e74642.
Huan T, Joehanes R, Schurmann C, Schramm K, Pilling LC, Peters MJ, et al. A whole-blood transcriptome meta-analysis identifies gene expression signatures of cigarette smoking. Hum Mol Genet. 2016;25:4611–23.
Junyent M, Tucker KL, Smith CE, Garcia-Rios A, Mattei J, Lai CQ, et al. The effects of ABCG5/G8 polymorphisms on plasma HDL cholesterol concentrations depend on smoking habit in the Boston Puerto Rican health study. J Lipid Res. 2009;50:565–73.
Dumitrescu L, Carty CL, Franceschini N, Hindorff LA, Cole SA, Bůžková P, et al. No evidence of interaction between known lipid-associated genetic variants and smoking in the multi-ethnic PAGE population. Hum Genet. 2013;132:1427–31.
The Airlie (VA) Consensus Conference. Standardization of Anthropometric Measurements. In: Lohman TRA, Roche A, Martorel R, editors. Champaign:Human Kinetics Publishers. 1988. p. 39–80.
Gaudet D, Arsenault S, Belanger C, Hudson T, Perron P, Bernard M, et al. Procedure to protect confidentiality of familial data in community genetics and genomic research. Clin Genet. 1999;55:259–64.
McNamara JR, Schaefer EJ. Automated enzymatic standardized lipid analyses for plasma and lipoprotein fractions. Clin Chim Acta. 1987;166:1–8.
Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502.
Vohl MC, Lepage P, Gaudet D, Brewer CG, Betard C, Perron P, et al. Molecular scanning of the human PPARα gene: association of the L162V mutation with hyperapobetalipoproteinemia. J Lipid Res. 2000;41:945–52.
Bijvoet SM, Hayden MR. Mismatch PCR: a rapid method to screen for the Pro207Leu mutation in the lipoprotein lipase (LPL) gene. Hum Mol Genet. 1992;1:541.
Monsalve MV, Henderson H, Roederer G, Julien P, Deeb S, Kastelein JJ, et al. A missense mutation at codon 188 of the human lipoprotein lipase gene is a frequent cause of lipoprotein lipase deficiency in persons of different ancestries. J Clin Invest. 1990;86:728–34.
De Bruin TW, Mailly F, van Barlingen HH, Fisher R, Castro Cabezas M, Talmud P, et al. Lipoprotein lipase gene mutations D9N and N291S in four pedigrees with familial combined hyperlipidaemia. Eur J Clin Investig. 1996;26:631–9.
Ma Y, Wilson BI, Bijvoet S, Henderson HE, Cramb E, Roederer G, et al. A missense mutation (Asp250Asn) in exon 6 of the human lipoprotein lipase gene causes chylomicronemia in patients of different ancestries. Genomics. 1992;13:649–53.
Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–8.
Barter PJ, Ballantyne CM, Carmena R, Castro Cabezas M, Chapman MJ, Couture P, de Graaf J, et al. ApoB versus cholesterol in estimating cardiovascular risk and in guiding therapy: report of the thirty-person/ten-country panel. J Intern Med. 2006;259:247–58.
Wilkins JT, Li RC, Sniderman A, Chan C, Lloyd-Jones DM. Discordance between Apolipoprotein B and LDL-cholesterol in young adults predicts coronary artery calcification: the CARDIA study. J Am Coll Cardiol. 2016;67:193–201.
Slagter SN, van Vliet-Ostaptchouk JV, Vonk JM, Boezen HM, Dullaart RP, Kobold AC, et al. Associations between smoking, components of metabolic syndrome and lipoprotein particle size. BMC Med. 2013;11:195.
Bentley AR, Sung YJ, Brown MR, Winkler TW, Kraja AT, Ntalla I, et al. Multi-ancestry genome-wide gene-smoking interaction study of 387,272 individuals identifies new loci associated with serum lipids. Nat Genet. 2019;51:636–48.
Prugger C, Wellmann J, Heidrich J, De Bacquer D, Perier MC, Empana JP, et al. Passive smoking and smoking cessation among patients with coronary heart disease across Europe: results from the EUROASPIRE III survey. Eur Heart J. 2014;35:590–8.
Prugger C, Wellmann J, Heidrich J, De Bacquer D, De Backer G, Périer MC, et al. Readiness for smoking cessation in coronary heart disease patients across Europe: results from the EUROASPIRE III survey. Eur J Prev Cardiol. 2015;22:1212–9.
Park KH, Shin DG, Cho KH. Dysfunctional lipoproteins from young smokers exacerbate cellular senescence and atherogenesis with smaller particle size and severe oxidation and glycation. Toxicol Sci. 2014;140:16–25.
Sniderman AD, Islam S, McQueen M, Pencina M, Furberg CD, Thanassoulis G, et al. Age and cardiovascular risk attributable to Apolipoprotein B, Low-Density Lipoprotein Cholesterol or Non-High-Density Lipoprotein Cholesterol. J Am Heart Assoc. 2016;5:e003665.
Niemiec P, Nowak T, Iwanicki T, Gorczynska-Kosiorz S, Balcerzyk A, Krauze J, et al. The rs2516839 polymorphism of the USF1 gene may modulate serum triglyceride levels in response to cigarette smoking. Int J Mol Sci. 2015;16:13203–16.
Freeman DJ, Caslake MJ, Griffin BA, Hinnie J, Tan CE, Watson TD, et al. The effect of smoking on post-heparin lipoprotein and hepatic lipase, cholesteryl ester transfer protein and lecithin:cholesterol acyl transferase activities in human plasma. Eur J Clin Investig. 1998;28:584–91.
Djousse L, Myers RH, Coon H, Arnett DK, Province MA, Ellison RC. Smoking influences the association between apolipoprotein E and lipids: the National Heart, Lung, and Blood Institute family heart study. Lipids. 2000;35:827–31.
Eliasson B, Mero N, Taskinen MR, Smith U. The insulin resistance syndrome and postprandial lipid intolerance in smokers. Atherosclerosis. 1997;129:79–88.
Kong C, Nimmo L, Elatrozy T, Anyaoku V, Hughes C, Robinson S, Richmond W, Elkeles RS. Smoking is associated with increased hepatic lipase activity, insulin resistance, dyslipidemia and early atherosclerosis in type 2 diabetes. Atherosclerosis. 2001;156:373–8.
Santamarina-Fojo S, González-Navarro H, Freeman L, Wagner E, Nong Z. Hepatic lipase, lipoprotein metabolism, and atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1750–4.
Ligthart S, Steenaard RV, Peters MJ, van Meurs JB, Sijbrands EJ, Uitterlinden AG, et al. Tobacco smoking is associated with DNA methylation of diabetes susceptibility genes. Diabetologia. 2016;59:998–1006.
Shifman S, Darvasi A. The value of isolated populations. Nat Genet. 2001;28:309–10.
Dallongeville J, Marecaux N, Fruchart JC, Amouyel P. Cigarette smoking is associated with unhealthy patterns of nutrient intake: a meta-analysis. J Nutr. 1998;128:1450–7.
Acknowledgements
The authors would like to acknowledge the contribution of the subjects who participated in this project as well as the staff of the ECOGENE-21 Biocluster and Chicoutimi Hospital Lipid Clinic for their dedicated work.
Funding
This study was supported by ECOGENE-21, whose contribution covered the cost associated with data collection and analysis.
Author information
Authors and Affiliations
Contributions
NR was involved in data analysis and has written themanuscript. DG has conceived the study, performed data collection and was a major contributor in writing the manuscript. GT was involved in the study conception as well as data collection and was a major contributor in writing the manuscript. DB was involved in the study conception, performed the data analysis and was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study has received the approval of the Chicoutimi Hospital Ethics Committee (2007–009 and 2007–010), and was conducted in accordance with the Declaration of Helsinki. Subjects gave a signed informed consent to participate in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Roy, N., Gaudet, D., Tremblay, G. et al. Association of common gene-smoking interactions with elevated plasma apolipoprotein B concentration. Lipids Health Dis 19, 98 (2020). https://doi.org/10.1186/s12944-020-01287-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-020-01287-7