
Abstract
Background
We previously identified the c.344G > A: p.(Arg115His) variant in the low-density lipoprotein receptor (LDLR) gene, which was interpreted as “conflicting interpretations of pathogenicity” in ClinVar, based on a genetic analysis of patients with familial hypercholesterolemia (FH). However, whether this variant affects the pathophysiology of FH remains unclear. Therefore, our aim was to annotate the c.344G > A: p.(Arg115His) variant in the LDLR gene in FH. We present 2 families harboring the c.344G > A: p.(Arg115His) variant in the LDLR gene.
Methods
Genetic analyses were performed for the coding regions and the exon-intron boundary sequence of the LDLR and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes in 2 FH families. Next, the family without pathogenic variants in the LDLR and PCSK9 genes was screened by whole-exome sequencing. Detailed clinical and biochemical data were gathered from family members.
Results
In one family, the index case had biallelic c.1567G > A: p.(Val523Met) and c.344G > A: p.(Arg115His) variants in the LDLR gene, while the sibling had only the c.1567G > A: p.(Val523Met) variant in the LDLR gene. There was no difference in the FH phenotype between the siblings. In another family, the index case and the sibling had no pathogenic variants in the LDLR, PCSK9, and apolipoprotein B (APOB) genes, but the sibling’s wife with nonFH had the c.344G > A: p.(Arg115His) variant in the LDLR gene. The sibling and his wife had 4 children, including an unaffected child and an affected child who had the c.344G > A: p.(Arg115His) variant in the LDLR gene. In addition, the allele frequency of the c.344G > A: p.(Arg115His) variant (0.0023–0.0043) in Japanese and East Asian populations is relatively high compared with that of the other LDLR pathogenic variants (0.0001–0.0008).
Conclusions
The c.344G > A: p.(Arg115His) variant in the LDLR gene is interpreted as benign in individuals with FH.
Similar content being viewed by others


Background
Familial hypercholesterolemia (FH) is a disease that leads to a high risk of coronary artery disease (CAD) because FH patients are exposed to high serum LDL-cholesterol (LDL-C) levels from birth. The prevalence of FH is 1 per 200–500 individuals in the general population [1]. FH is caused by mutations in the low-density lipoprotein receptor (LDLR) and in the related genes apolipoprotein B (APOB) and proprotein convertase subtilisin/kexin type 9 (PCSK9). More than 4970 variants in the LDLR gene, 580 variants in the APOB gene, and 350 variants in the PCSK9 gene are shown in ClinVar [2]. In Japan, pathogenic variants in the APOB gene have not been reported [3]. Whether some variants of the LDLR and PCSK9 genes affect the pathophysiology of FH remains unclear.
In our FH cohort, patients were found to harbor the c.344G > A: p.(Arg115His) variant in the LDLR gene, which was interpreted as “conflicting interpretations of pathogenicity” in ClinVar. However, whether this variant affects the pathophysiology of FH remains to be elucidated. Herein, we present 2 families harboring the c.344G > A: p.(Arg115His) variant in the LDLR gene, including an index case with biallelic LDLR variants that comprised the c.344G > A: p.(Arg115His) and a pathogenic variant (Family 1) and nonFH patients with the c.344G > A: p.(Arg115His) variant (Family 2).
Methods
The protocol of this study was approved by the Ethics Review Committee of the National Cerebral and Cardiovascular Center (M17–56 or M24–80). Each patient provided written informed consent to participate in the study.
DNA analysis
Genomic DNA was extracted from whole blood from the sibling of Family 1 and the index case of Family 2 using an automated DNA extraction machine (QIAsymphony; QIAGEN, Valencia, CA) and from the other members of Family 2 using the Genome Extraction Kit (GENOMIX; Biologica, Nagoya, JAPAN) by SRL Inc. (Tokyo, JAPAN). The coding regions and exon-intron boundary sequences of the LDLR and PCSK9 genes were examined by Sanger sequencing as described previously [4]. For the index case of Family 2, multiplex ligation-dependent probe amplification (MLPA) was performed to detect large rearrangements of the LDLR gene using the P062B LDLR MLPA Kit (MRC Holland, Amsterdam, the Netherlands). Next, whole-exome sequencing was performed for Family 2. Exome libraries were prepared using the SureSelect Human All Exon V7 Kit (Agilent Technologies, Santa Clara, CA). Sequencing was performed by NovaSeq 6000 (Illumina, San Diego, CA) with 150 bp paired-end reads at RIKEN GENESIS CO., LTD. Sequence reads were aligned to the human reference genome (hg19) using BWA-MEM. Single nucleotide variants and small indels were called with HaplotypeCaller of the Genome Analysis Tool Kit. The presence/absence of the c.344G > A:p.(Arg115His) variant in the LDLR gene was confirmed by Sanger sequencing.
Clinical and laboratory data
Serum total cholesterol (TC), triglycerides (TG), and high-density lipoprotein-cholesterol (HDL-C) levels were measured using enzymatic methods. LDL-C levels were calculated by the Friedewald formula or were measured using enzymatic methods. Achilles tendon thickness (ATT) was measured by X-ray. CAD was evaluated by the presence of myocardial infarction, angina pectoris, or coronary arteries with ≥ 75% stenosis by coronary angiography or electrocardiogram.
Results
Case presentation
Family 1
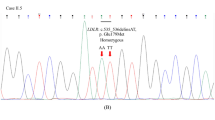
The index case was a 19-year-old woman who had biallelic LDLR c.1567G > A: p.(Val523Met) and c.344G > A: p.(Arg115His) variants (Fig. 1, Table 1). She was referred to our lipid clinic with her brother at the age of 19 years for dyslipidemia, and her untreated LDL-C level was 208 mg/dL. She did not have ATT. Her medications included 2.5 mg of rosuvastatin, and her LDL-C level was 100 mg/dL on this medication regimen. Her sibling (II-1) was a 21-year-old male who had the LDLR c.1567G > A: p.(Val523Met) variant. His untreated LDL-C level was 204 mg/dL at 21 years old. His ATT values were 6.7 and 9.1 mm. His medication included 2.5 mg of rosuvastatin, and his LDL-C level was 99 mg/dL under this medication regimen. Their father was diagnosed with FH, but his lipid profile was unknown.

Two family pedigrees with the c.344G > A: p.(Arg115His) variant in the LDLR gene. Arrows show the index cases
Family 2
The index case was a 52-year-old man who had no pathogenic variants in the LDLR and PCSK9 genes (Fig. 1). He was referred to the lipid clinic at the age of 52 years for dyslipidemia (II-1). His untreated LDL-C level was 237 mg/dL, and his ATT values were 7.1 mm and 9.2 mm. He was diagnosed with hypertension at the age of 45 years. His medication regimen included 5 mg of rosuvastatin and 10 mg of ezetimibe, and his LDL-C level was 111 mg/dL under this regimen. The lipid profiles of his parents were unknown. His father died of liver cancer at 65 years old, and his mother died at 64 years old due to unknown causes. The sibling (II-2) of the index case was a 49-year-old man who was diagnosed with FH. His medication regimen included 2 mg of rosuvastatin, 10 mg of ezetimibe, and 2 g of omega-3 fatty acid ethyl, and his LDL-C level was 106 mg/dL under this regimen. The index case and the sibling’s family were screened by whole-exome sequencing. We examined the coding regions and exon-intron boundary sequences in the LDLR, PCSK9, and APOB genes. The index case and the sibling had no pathogenic variants in the LDLR, PCSK9, and APOB genes, but the wife of the sibling (II-3) with nonFH had the c.344G > A: p.(Arg115His) variant in the LDLR gene. They had no loss-of-function mutations in the PCSK9 and APOB genes. The sibling and the wife had 4 children, of whom 3 siblings (III-1, III-2, and III-3) were diagnosed with nonFH and 1 sibling (III-4) was diagnosed with FH. The eldest daughter (III-1) and the youngest son (III-4) were heterozygous for the c.344G > A: p.(Arg115His) variant in the LDLR gene. The presence/absence of the c.344G > A:p.(Arg115His) variant in the LDLR gene was confirmed by Sanger sequencing.
Allele frequency of the c.344G > A: p.(Arg115His) variant in the LDLR gene in Japanese and East Asian populations.
The allele frequency of the c.344G > A: p.(Arg115His) variant in the LDLR gene in Japanese and East Asian populations was 0.0029, 0.0043, and 0.0023 according to the Japanese Human Genetic Variation Database (n = 1201; HGVD: http://www.genome.med.kyoto-u.ac.jp/SnpDB) [5], the Tohoku Medical Megabank Organization database (n = 3381; tommo_3.5KJPN) [6], and ExAC (n = 66,000, [7]), respectively. The allele frequency of the c.344G > A: p.(Arg115His) variant in the LDLR gene was relatively high compared with that of the other LDLR pathogenic variants (0.0001–0.0008) detected in these databases.
Discussion
We present 2 families harboring the c.344G > A: p.(Arg115His) variant in the LDLR gene. In the first family (Family 1), we identified an index case of biallelic LDLR variants that included c.1567G > A: p.(Val523Met) and c.344G > A: p.(Arg115His), and in the second family (Family 2), we identified that the wife of the index case’s sibling with FH, who was normolipidemic, had the LDLR c.344G > A: p.(Arg115His) variant. In Family 1, the index case had biallelic LDLR variants that included c.1567G > A: p.(Val523Met) and c.344G > A: p.(Arg115His), while the sibling had only the heterozygous c.1567G > A: p.(Val523Met) variant in the LDLR gene. The c.1567G > A: p.(Val523Met) variant is defined as pathogenic/likely pathogenic in ClinVar. FH patients harboring biallelic LDLR pathogenic variants generally show a much more severe phenotype than those harboring heterozygous LDLR pathogenic variants. However, the phenotype of the index case in Family 1 was identical to that of heterozygous FH. In Family 2, the index case’s sibling with FH had 4 children, namely, 1 affected and 3 unaffected siblings. His wife with nonFH had the c.344G > A: p.(Arg115His) variant in the LDLR gene. A heterozygous c.344G > A: p.(Arg115His) variant in the LDLR gene was detected in 1 unaffected sibling and 1 affected sibling. The FH phenotype of the youngest son is suggested to be derived from his father harboring unknown pathogenic variants.
The c.344G > A: p.(Arg115His) variant in the LDLR gene has been reported in several individuals of Asian ethnicity [3, 8]. The allele frequency of the c.344G > A: p.(Arg115His) (0.029/0.0043) variant in the LDLR gene among the Japanese population was similar to that expected based on the prevalence of heterozygous FH among the Japanese population (0.002–0.005). The allele frequency of the c.344G > A: p.(Arg115His) variant in the LDLR gene is relatively high compared with that of other LDLR pathogenic variants (0.0001–0.0008) in Japanese databases. The LDLR gene is located on chromosome 19p13.1–13.3 and contains 18 exons, encoding a mature protein of 839 amino acids with a 21 amino acid signal peptide. The c.344G > A: p.(Arg115His) variant is located in the cysteine-rich, 40 amino acid repeat region of the binding domain of the LDLR, but the 115th Arg is not conserved among this region [9]. In addition, the Arg (pI = 10.76) to His (pI = 7.59) substitution does not change the positive charge of this sequence. For the c.344G > A: p.(Arg115His) variant, only one study exists; functional assay results revealed that receptor activities were 64% of normal in COS7 cells transfected with LDLR cDNA containing the c.344G > A: p.(Arg115His) variant [10]. However, the genotype-phenotype correlation of the c.344G > A:p.(Arg115His) in the LDLR gene in pedigrees has not been demonstrated. Thus, based on 2 pedigree-based genetic analyses, the c.344G > A: p.(Arg115His) variant in the LDLR gene was classified as benign according to the guidelines issued by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Thus, examining the genotype-phenotype correlation in a pedigree is important for providing a genetic diagnosis with high accuracy.
In our cohort, we showed that unrelated patients harboring no pathogenic variants in the LDLR and PCSK9 genes comprised approximately 40% of FH patients [4]. FH may be partly explained by the accumulation of common SNPs [11]. A number of factors, including somatic genetic changes, environmental factors, and other genetic factors, might contribute to the pathogenesis and phenotypic variations observed in FH. We are currently searching for new candidate FH genes that may be responsible for FH in Family 2. Finally, in vitro functional analyses are needed to quantify the actual effect of the c.344G > A: p.(Arg115His) variant in the LDLR gene.
Conclusions
The c.344G > A: p.(Arg115His) variant in the LDLR gene was not shared by the sibling with FH in Family 1, and patients with nonFH also had the variant in Family 2. In conclusion, the c.344G > A: p.(Arg115His) variant in the LDLR gene is interpreted as benign based on pedigree-based genetic analysis.
Availability of data and materials
The data are not available because some data are being used by another study.
Abbreviations
- APOB:
-
Apolipoprotein B
- ATT:
-
Achilles tendon thickness
- CAD:
-
Coronary artery disease
- FH:
-
Familial hypercholesterolemia
- HDL:
-
High-density lipoprotein
- HDL-C:
-
HDL-cholesterol
- LDL:
-
Low-density lipoprotein
- LDL-C:
-
LDL-cholesterol
- LDLR:
-
LDL receptor
- MLPA:
-
Multiplex ligation-dependent probe amplification
- PCSK9:
-
Proprotein convertase subtilisin/kexin type 9
- TC:
-
Total cholesterol
- TG:
-
Triglycerides
References
Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri MA, Tada H, et al. Molecular genetic epidemiology of homozygous familial hypercholesterolemia in the Hokuriku district of Japan. Atherosclerosis. 2011;214:404–7.
Iacocca MA, Chora JR, Carrie A, Freiberger T, Leigh SE, Defesche JC, et al. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum Mutat. 2018;39:1631–40.
Yu W, Nohara A, Higashikata T, Lu H, Inazu A, Mabuchi H. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis. 2002;165:335–42.
Hori M, Ohta N, Takahashi A, Masuda H, Isoda R, Yamamoto S, et al. Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients. Atherosclerosis. 2019;289:101–8.
Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547–53.
Nagasaki M, Yasuda J, Katsuoka F, Nariai N, Kojima K, Kawai Y, et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun. 2015;6:8018.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Kim JH, Choi HK, Lee H, Park HY, Kim JH, Kim JW, et al. Novel and recurrent mutations of the LDL receptor gene in Korean patients with familial hypercholesterolemia. Mol Cells. 2004;18:63–70.
Sudhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science. 1985;228:815–22.
Chang JH, Pan JP, Tai DY, Huang AC, Li PH, Ho HL, et al. Identification and characterization of LDL receptor gene mutations in hyperlipidemic Chinese. J Lipid Res. 2003;44:1850–8.
Talmud PJ, Shah S, Whittall R, Futema M, Howard P, Cooper JA, et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet. 2013;381:1293–301.
Acknowledgments
We thank Dr. Yoshihiro Miyamoto, Mr. Suguru Yamamoto, Mr. Naotaka Ohta, Mr. Hiroaki Masuda, and Ms. Rieko Isoda for DNA analysis and Ms. Rie Oishi for clerical support.
Funding
This work was partly supported by grants from the Ministry of Health, Labor and Welfare of Japan for Clinical Research on Intractable Diseases (H26-nanji-ippan-056, H30-nanji-ippan-003), JSPS KAKENHI Grant Number JP17K08681, the Japan Agency for Medical Research and Development (16ek0210075h0001), the Intramural Research Fund (28–2-2; 29–6-11) for Cardiovascular Diseases of the National Cerebral and Cardiovascular Center, the Japan Heart Foundation & Astellas Grant for Research on Atherosclerosis Update, the Takeda Science Foundation, the Kanae Foundation for the Promotion of Medical Science, Novartis Research Grants, and The Japanese Circulation Society.
Author information
Authors and Affiliations
Contributions
M.H. collected the clinical data, and M.H., M.O. and M.H-S. contributed to the interpretation of the data. A.T. performed bioinformatic analysis in whole-exome sequencing. M.H. and C. S performed the genetic analysis by Sanger sequencing. M.H. drafted the manuscript and contributed to the conception and design of the study. M.H-S. contributed to the study’s supervision. All authors gave final approval of the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The protocol of this study was approved by the Ethics Review Committee of the National Cerebral and Cardiovascular Center (M17–56 or M24–80). Each patient provided written informed consent to participate in the study.
Consent for publication
All the participants provided written informed consent for the publication of the results of this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hori, M., Takahashi, A., Son, C. et al. The benign c.344G > A: p.(Arg115His) variant in the LDLR gene interpreted from a pedigree-based genetic analysis of familial hypercholesterolemia. Lipids Health Dis 19, 62 (2020). https://doi.org/10.1186/s12944-020-01252-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-020-01252-4