
Abstract
Non coding RNAs (ncRNAs) have emerged as regulators of human carcinogenesis by affecting the expression of key tumor suppressor genes and oncogenes. They are divided into short and long ncRNAs, according to their length. Circular RNAs (circRNAs) are included in the second group and were recently discovered as being originated by back-splicing, joining either single or multiple exons, or exons with retained introns. The human Plasmacytoma Variant Translocation 1 (PVT1) gene maps on the long arm of chromosome 8 (8q24) and encodes for 52 ncRNAs variants, including 26 linear and 26 circular isoforms, and 6 microRNAs. PVT1 genomic locus is 54 Kb downstream to MYC and several interactions have been described among these two genes, including a feedback regulatory mechanism. MYC-independent functions of PVT1/circPVT1 have been also reported, especially in the regulation of immune responses. We here review and discuss the role of both PVT1 and circPVT1 in the hematopoietic system. No information is currently available concerning their transforming ability in hematopoietic cells. However, present literature supports their cooperation with a more aggressive and/or undifferentiated cell phenotype, thus contributing to cancer progression. PVT1/circPVT1 upregulation through genomic amplification or rearrangements and/or increased transcription, provides a proliferative advantage to malignant cells in acute myeloid leukemia, acute promyelocytic leukemia, Burkitt lymphoma, multiple myeloma (linear PVT1) and acute lymphoblastic leukemia (circPVT1). In addition, PVT1 and circPVT1 regulate immune responses: the overexpression of the linear form in myeloid derived suppressor cells induced immune tolerance in preclinical tumor models and circPVT1 showed immunosuppressive properties in myeloid and lymphoid cell subsets. Overall, these recent data on PVT1 and circPVT1 functions in hematological malignancies and immune responses reflect two faces of the same coin: involvement in cancer progression by promoting a more aggressive phenotype of malignant cells and negative regulation of the immune system as a novel potential therapy-resistance mechanism.
Similar content being viewed by others


Background
Non-coding RNAs (ncRNAs) are transcripts that do not encode proteins. They are diffused in the human genome and dysregulated in cancer cells. Given that genes for ncRNAs are often located in fragile sites (FRA), in regions with loss of heterozygosity and common breakpoint sites, they represent a new class of transcripts that participates in tumorigenesis [1, 2]. Some ncRNAs have a tumor suppressor function while others act as oncogenes [3, 4]. ncRNAs are classified into two categories on the basis of the length of their sequence: short ncRNAs do not exceed 200 nucleotides in size, while long ncRNAs (lncRNAs) are characterized by longer sequences. Short ncRNAs have been extensively studied. However, lncRNAs remain largely explored.
LncRNAs generally have a 5′ terminal methylguanosine cap, are frequently polyadenylated and alternatively spliced [5, 6]. They have a thermodynamically stable secondary structure with hairpin loops and bulges [7], that enables them to interact with DNA, mRNAs, ncRNAs and proteins. lncRNAs regulate gene expression at different levels, from mRNA translation to cytoplasmatic and nuclear epigenetic processes, including miRNA sponging [8].
Circular RNAs (circRNAs) represent an emerging group of cellular lncRNAs. They are covalently closed loop-like structure with no 5′ and 3′ polarity and this circular structure confers them an increased stability and resistance to the cellular linear RNA decay machineries [9]. Evidence suggests that circRNAs have an independent biogenesis, which is unrelated to canonical splicing of linear RNA. Indeed they result from a back-splicing of the 5′ splice position with the 3′ splice position, or from exon skipping [10]. circRNAs expression is partially regulated by DNA methylation of host genes. In particular, a recent study showed that knockdown of DNMT3A induces circRNAs expression in a host gene dependent manner, while changes in their level is largely independent of host gene regulation upon silencing of DNMT3B [11]. Once synthesized, circRNAs tend to form 16–26 base pair intra-molecularly imperfect RNA duplexes and can be degraded by RNase L upon activation of early innate immune response [12].
Human Plasmacytoma Variant Translocation 1 (PVT1) gene is located on chromosome band 8q24.21 (Fig. 1) and encodes for both circRNAs and linear ncRNA isoforms, as well as 6 microRNAs. In this review, we describe the currently available data on the role of both PVT1 and circPVT1 in hematopoietic cells and especially in hematological malignancies, including genomic alterations, involvement in disease progression and in the regulation of the immune response, which represents a potential therapy-resistance mechanism.

PVT1 transcript alignment in UCSC Genome Browser on Human Dec. 2013 (GRCh38/hg38) Assembly. 8q24.21 region encodes for PVT1 and circPVT1. The sequence of PVT1 transcript with the largest number of exons is shown in the upper part of the figure. PVT1 isoforms and circPVT1 with the highest expression in the hematopoietic system, are shown in the lower part of the figure (from the top: NONHSAT255418.1, NONHSAT255436.1, NONHSAT255445.1). The secondary structure of circPVT1 is reported as discovered by Liu et al. [12]. The rectangles represent the exons
PVT1 and circPVT1: different isoforms with a different regulation
Twenty-six circPVT1 isoforms have been annotated in the CircInteractome Database (https://circinteractome.nia.nih.gov/index.html) [13], with a spliced length ranging from 113 to 11,130; 8 of them were also detected in the K562 and Gm12878 hematopoietic models, among others. The most common isoform is 410 bp. It is a product of back-splicing and contains the whole exon 2 of PVT1, forming a closed loop-like structure [14, 15]. Conversely, PVT1 exists in 26 different transcript variants (www.noncode.org) [16], with some of them not containing exon 2. The different variants are capped at the 5′ and polyadenylated at 3′ end [17]. PVT1 isoforms are variably expressed across human tissues, with adrenal gland and heart displaying the highest expression (www.noncode.org). Hematopoietic tissues, namely lymph node and white blood cells, showed high levels of few isoforms (Fig. 1), while most of them were barely detectable (Fig. 2a).

PVT1 expression in the hematopoietic system. aPVT1 isoforms detected in lymph node and/or white blood cells (www.noncode.org). FPKM from Illumina’s Human BodyMap 2.0 project are shown (http://www.ensembl.info/2011/05/24/human-bodymap-2-0-data-from-illumina/). b Overall PVT1 expression in hematopoietic cell populations (GSE98791). Data from Agilent-021441 NCode Human Long Non-coding RNA microarray were analyzed with Feature Extraction Software10.5 (Agilent) [18]. The processed signal intensity of PVT1 is represented in the figure (HSC: hematopoietic stem cells, ET: in vitro-differentiated erythroblasts, MK: in vitro-derived megakaryocytes, GR: granulocytes, MONO: monocytes, B: B lymphocytes, NK: natural killer cells, CD4 + T: CD4+ T lymphocytes, CD8 + T: CD8+ T lymphocytes)
PVT1 and circPVT1 are transcribed from two different promoters, thus confirming an independent regulation of their expression, with circPVT1 promoter being upstream of exon 2 [19]. Accordingly, Chen et al. reported that the expression levels of PVT1 and circPVT1 were poorly correlated in gastric cancer tissue and human gastric epithelium GES-1 line [20]. Moreover, an independent post-transcriptional regulation of PVT1 and circPVT1 has been suggested on the basis of their structure and localization. circPVT1 mostly localizes in the cytoplasm, while PVT1 is primarily nuclear [21]. In addition, thanks to its circular structure, circPVT1 is resistant to exonuclease activity of RNase R and it was suggested to form a protein-coding open reading frame (ORF) of 104 amino acids [22], whose expression and role across cancer deserves further investigation. Regarding their function, both isoforms are upregulated in many cancer types and correlated with various clinical features, including overall survival and lymph node metastases [23]. However, these associations may vary among cancer types and the specific PVT1 isoform (linear or circular), clearly suggesting the need to disentangle their biological function at cellular level [24].
The majority of studies focuses on the linear isoform of PVT1, that promotes cell growth and proliferation in cancer, as well as cell migration, invasion, and drug resistance. Moreover, a role as a sponge for tumor-suppressor miRNAs with oncogenic properties has been described for both PVT1 isoforms, even though the literature points on different miRNA entities, depending on cancer type, especially for circPVT1. Their pro-tumorigenic role is often attributed to their functional interaction with the MYC oncogene, localized about 54 Kb upstream PVT1. However, recent findings provided new insights on potential MYC-independent functions, especially for circPVT1, which will be addressed in the following sections.
Role of PVT1 and circPVT1 in the immune system
LncRNAs and circRNAs have been reported to participate in the differentiation and functioning of immune cells under physiological [25] and pathological conditions [26]. In the hematopoietic lineage, PVT1 is expressed in CD34+CD38− cord blood-derived stem cells [18] (GSE98791, Fig. 2b). Moreover, in mature cells, high levels of this transcript were detected in peripheral blood (PB) T lymphocytes (CD4+ > CD8+) and, mostly, in vitro-differentiated erythroblasts (Fig. 2b). Accordingly, Gillinder et al. reported an increase in PVT1 transcription early after erythropoietin (EPO) stimulation in the murine immature erythroid J2E cell line [27], that proliferates and terminally differentiates following EPO exposure. However, the role of PVT1 in the erythroid lineage has not been further elucidated.
PVT1 expression, including extracellular and intracellular RNA, increased in the PB of mice 16, 24 and 48 h after whole body irradiation (2–8 Gy) [28], along with other p53-related genes. Since radiation leads to DNA damage, with consequent activation of the p53-dependent DNA repair pathways and since p53 binding to its responsive element contributes to PVT1 upregulation [29], PVT1 may serve as a biomarker of DNA damage response. Therefore, PVT1 might be potentially translated to the clinics for an early evaluation of ablative regimens or as an easy-to-use readout of activation of a DNA damage response.
In parallel, preclinical and clinical data suggest a potential role for PVT1 and/or circPVT1 in the myeloid and lymphoid lineages. From a structural point of view, a genome wide association study identified 44 variants in the PVT1 gene correlating with selective IgA deficiency [30]. The peak PVT1 variant was in moderate linkage disequilibrium with four variants with a potential regulatory role (rs1499364, rs7001706, rs35135218, rs10601187) that were predicted to affect binding and were located in transcription factors binding sites. In particular, rs1499364 lies in a region of open chromatin and in a histone mark for active transcription in regulatory T (Treg) cells. Moreover, the intronic variant rs7001706 is located in a H3K4me1 histone mark in Treg cells and in a FOXP3 transcription factor binding motif. Of note, IgA deficiency, which results in defective regulation of mucosal immunity and gut commensalism, with recurrent mucosal infections [31, 32], shows a higher penetrance in families with autoimmunity recurrence. In particular, the prevalence of systemic lupus erythematosus (SLE), type 1 diabetes and celiac disease are respectively 10 and 35 times higher in patients affected by IgA deficiency, compared with the general population [33].
At functional level, circPVT1 is significantly reduced in monocytes, B and T lymphocytes from the PB of SLE patients, along with other circRNAs having intra-double stranded (ds) RNA duplexes, while the expression of their linear cognate mRNAs is marginally affected [12]. The reduced level of circRNAs in SLE patients has been linked to the spontaneous activation of RNase L [12], a cytoplasmic endoribonuclease that is generally activated by pathogenic dsRNAs or viral infection [34]. RNase L-mediated degradation of circRNAs, including circPVT1, has been also demonstrated in acute T cell leukemia and acute monocytic leukemia cellular model and is responsible for hyperactivation of the interferon (IFN)-inducible serine/threonine protein kinase PKR, that physiologically occurs in the early stage of the innate immune responses [12]. Exogenous expression of circRNAs reduced PKR activation and EIF2α phosphorylation in T cells from SLE patients and suppressed IFN-β and type I IFN-induced gene signatures, which are hallmarks of SLE. These data indicate a role of circPVT1 in the regulation of immune responses and the possibility of using circRNAs as potential therapies against autoimmune diseases. In addition, reduced expression of PVT1 was reported in PB cells of relapsing-remitting multiple sclerosis patients compared with healthy subjects [35] and a role for PVT1 has been reported in the inflammatory processes involved in asthma [36] and septic acute kidney injury [37].
While evidence is available through the literature regarding the immunoregulatory role of PVT1 and/or circPVT1 under infection, inflammatory conditions and autoimmune disease, little is known about their function in the immune system during malignant transformation. High expression of linear PVT1 has been detected in granulocytic myeloid-derived suppressor cells (G-MDSC) from tumor tissues in murine models of Lewis lung carcinoma and colorectal cancer [38]. PVT1 expression was induced by HIF-1α in tumor-infiltrating G-MDSC, which experiment hypoxic conditions. In these cells, PVT1 regulates ARG1 activity and reactive oxygen species (ROS) production, thus contributing to the suppression of T-cell-induced antitumor immune responses. Indeed, the proportion of CD4+ IFN-γ+ T helper 1 and CD8+ IFN-γ+ cytotoxic T lymphocytes was increased in tumor tissues (the latter also in the draining lymph nodes) of mice injected with G-MDSC lacking PVT1 expression.
These data on the MDSC-mediated function of PVT1 suggest that its overexpression in specific immune cell subsets has the capability of dampening anti-tumor responses and potentially contribute to therapy resistance, especially in the therapeutic settings relying on immune cell reactivation (e.g. immune checkpoints-based regimens, which are largely exploited for combination strategies). Moreover, the observation that PVT1 is highly expressed in the T cell lineage and that circPVT1 levels are reduced in lymphocytes from SLE patients suggest novel potential direct implications of either the linear and the circular isoform in the T cell-mediated anti-tumor response.
PVT1 and circPVT1 in hematological malignancies
Chromosome 8q24.21 is a target for genomic rearrangement across cancer, including hematological malignancies. Particularly, it is frequently involved in the emergence of aberrant chimeric genes, high copy number gains, both of them associated with poor prognosis in human cancers. Moreover, it harbors a number of susceptibility loci at 8q24.21 near or in the PVT1 gene, such as single nucleotide variants in different types of lymphoma (Table 1).
Acute myeloid leukemia
Chromosomal rearrangements and copy number changes at the 8q24.21 locus are relatively frequent events in acute myeloid leukemia (AML) and play a role in its pathogenesis. Retroviral insertion analysis from various non-T cell derived mouse tumors identified the first integration at the pvt1 locus in myelogenous leukemia [71]. The first evidence of PVT1 involvement in human AML came from cytogenetic studies on the 8q24 locus and on double minutes chromosomes (dmin) [39]. Indeed, a 4.3 Mb minimal common amplicon was identified in MYC-containing dmin, with clustered distal breakpoints located downstream the PVT1 gene, among others [40]. A t(6;8)(p21;q24) translocation involving the TATA-binding protein-associated factor SUPT3H and PVT1 gene has been recently reported in blastic plasmacytoid dendritic cell neoplasm, with breakpoint regions mapping in exon 3 and exon 1, respectively [72]. In AML, PVT1 appeared as a breakpoint hotspot in MYC amplification. Indeed, 92% of AML cases carrying MYC amplifications as dmin, homogeneously staining region (hsr), or ring chromosomes (AML-amp) were characterized by expression of chimeric transcripts that frequently involved PVT1 as either a 5′ or 3′ partner, with PVT1 amplification. PVT1 fusion genes were generated as post-transcriptional events, since they were not identified at genomic level and showed a conserved breakpoint position (in the majority of cases) and MYC, FAM49B, RP11-89 K10, CCDC26, CASC11, and CASC8 as recurrent partners, with a predicted dysregulation of their protein product due to promoter swapping, loss of the untranslated region or N/C-terminus truncation, in case of protein coding genes as partners in fusions. For chimeras joining PVT1 with other non-coding transcripts, the role is presently unclear. The PVT1-CCDC26 and PVT1-NSMCE2 chimeras were also detected in AML cell lines [41] and primary cells [42], respectively, and PVT1-NSMCE2-rearranged AML showed amplification of both genes and relocation in micronuclei [42]. PVT1 and NSMCE2 overexpression and involvement in chimeric transcripts were also specific features of AML-amp cases with the highest numbers of chimeras. AML-amp cases carrying PVT1 amplification showed increased levels of PVT1 and circPVT1, the latter being also confirmed in AML cell lines with more than 5 copies of PVT1 [41].
Conversely, in the general AML population, controversial results have been reported regarding PVT1 expression levels in bone marrow (BM) blasts compared with healthy donors [43, 45, 73]. However, different groups agreed on the increased PVT1 level in acute promyelocytic leukemia (APL), compared with mononuclear cells [45] or granulocytes [46] from healthy donors. Genomic amplification of the 8q24 chromosomal region is a common secondary event in human APL [47], indicating that PVT1 may be involved in APL progression. PVT1 expression was downregulated, along with MYC, by all-trans retinoic acid (ATRA) in APL models, thus indicating a potential role for the lncRNA in ATRA-induced granulocytic differentiation [74] and cell cycle arrest [46]. MYC silencing was sufficient per se to reduce PVT1 levels. In turn, PVT1 knockdown resulted in decreased MYC protein, with no changes at mRNA level, clearly showing a dual relationship between PVT1 and MYC, the former controlling MYC protein synthesis and/or stability in malignant promyelocytes. This evidence suggests that PVT1 contributes to an aggressive phenotype in APL, by modulating cell proliferation. What comes first, whether MYC or PVT1, remains unresolved. The role of PVT1 in APL progression is also supported by its upregulation in high risk APL (white blood cell count> 10,000/mL) compared with intermediate and low risk cases (defined according to white blood cell and platelet count) [45]. A prognostic or functional role of PVT1 has also been reported in other AML subtypes. AML harbouring the t (8;21) translocation have higher PVT1 compared with other AML. In particular, high PVT1, or high MYC predicted shorter leukemia-free survival in t(8;21) AML patients [43], which suggested a potential mechanism of chemotherapy resistance in cells with leukemia initiating capacity, that drives disease progression. This hypothesis would reinforce the observation that PVT1 knockdown reverses multidrug resistance in solid tumors [75]. In line with its elevated level in normal erythroid cells, PVT1 was highly expressed in acute erythroleukemia (AEL) models [48]. Its inhibition led to a significant decrease of cell proliferation, with accumulation of cells in the G0/G1 phase of the cell cycle, that associated with downregulation of MYC protein and upregulation of p15 and p16 [48] (Fig. 3). BCL2 was also targeted by PVT1 silencing, thus resulting in induction of apoptosis and necrosis [17, 48, 49]. Consistently with the results obtained in APL, in the murine MLL-AF9/NRASG12D and MLL-ENL AML models, PVT1 depletion activated a myeloid differentiation program, with downregulation of cKit and leukemia stem cell signatures and upregulation of CD11b [44]. This phenotype resembled the one induced by bromodomain and extra-terminal domain (BET) protein inhibitors in AML [76] and was mediated by MYC downregulation, since its ectopic expression reversed the differentiation and anti-proliferative programs promoted by PVT1 silencing [44].

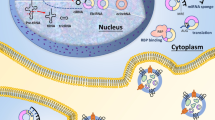
Model of PVT1 and circPVT1 pro-tumorigentic functions in hematological malignancies. PVT1 and circ-RNA act as a miRNA sponge and regulate the energy metabolism, protein stability, cell cycle progression through a variety of pathways, thus promoting cell proliferation, immune suppression and inhibiting cell death (green represents induction, red indicates suppression, dashed line shows indirect and not specified mechanisms; ROS: reactive oxygen species; RBPs: RNA-binding proteins, including EIF4A3, U2AF65, AGO2, AUF1, DGCR8, FUS, HNRNPC, PTB, TAF15, TDP43, TIA1, TIAL1, LIN28A)
Acute lymphoblastic leukemia
Acute lymphoblastic leukemia (ALL) is the only hematological malignancy in which a functional role of circPVT1 has been clearly demonstrated. Indeed, circPVT1 (but not PVT1) was specifically overexpressed in primary BM cells from B- and T-ALL, compared with healthy controls [50]. Notably, higher levels of the transcript were detected in younger patients (aged less than 35 years). Moreover, higher circPVT1 levels were detected in pediatric B-precursor ALL patient-derived xenograft samples, and in particular in ETV6-RUNX1 rearranged cases, compared with B cells from healthy donors [51]. Downregulation of circPVT1 in B- and T-ALL models had no effect on PVT1 (as also observed in gastric cancer) [20], while causing a significant reduction in the proliferation rate and induction of apoptosis, associated with a decrease of MYC and BCL2 protein expression [50] (Fig. 3). The observations that circPVT1 primarily localizes in the cytosol strongly support a role as miRNA sponge [77], reinforcing the hypothesis of a post-transcriptional regulation of circPVT1 over its target genes [50]. In particular, circPVT1 seemed to sponge let-7 and mir-125, that are known to modulate MYC and BCL-2, respectively [52] (Fig. 3). In parallel, PVT1 knockdown in the Jurkat T-ALL cell line also suppressed cell proliferation, with cell cycle arrest in G0/G1 phase, increased apoptosis [52] and upregulation of Caspase-3, p15, and p16 expression (Fig. 3). BCL2 and MYC were downregulated by PVT1 knockdown in T-ALL cells, as observed upon circPVT1 silencing [50] in B- and T-ALL (Fig. 3). These results may suggest a similar, but not compensatory, function of PVT1 and circPVT1 in ALL. However, we cannot exclude that downregulation of circPVT1, potentially occurring during PVT1 silencing, might be the driver of the observed phenotype. Specific experimental and cell engineering strategies will be required to clarify this question.
Chronic lymphocytic leukemia
Differently from other hematological malignancies, 8q24 rearrangements are generally rare (3.7% of amplified cases [78]) in B-cell chronic lymphocytic leukemia (CLL) and they are often acquired during the disease course [79]. Gain of 8q24 associates with poor overall survival and/or shorter time to first treatment [78, 80, 81] and is frequently detected in 17p-deleted CLL, where it has a negative prognostic value [82]. PVT1 has been investigated in a single CLL case with complex karyotype, t(8;13)(q24;q14) translocation and a deletion on the derivative chromosome 8 mapping downstream the MYC oncogene and encompassing the PVT1 locus [53]. Nevertheless, PVT1 was significantly upregulated, as well as MYC, as a consequence of the t(8;13) translocation and might have a potential role in the aggressive phenotype of that CLL case.
Lymphomas
The first PVT1 alterations identified in lymphoma refer to Burkitt lymphoma (BL) cases carrying the t(2;8) or t(8;22) translocations (~ 20%), that juxtapose the IGL or IGΚ locus with the PVT1 gene, resulting in chimeric transcripts that contain PVT1 exons 1a or 1b spliced to the IG light chain constant region [54,55,56]. PVT1 was recently reported as a mutational hotspot in endemic and sporadic BL [57]. Its downregulation in the BL Raji model decreased MYC protein expression and suppressed proliferation by promoting cell cycle arrest in the G0/G1 phase [58]. Accordingly, cell cycle-and DNA damage response-related genes were altered by PVT1 knockdown, including upregulation of CDKN2A, CDN1B, RB1, CCND2, GADD45A and downregulation of CDC20, CDK4, CD6, ATM and BRCA2.
Genomic events targeting the PVT1 locus have been also described in other lymphoma types. Pvt1 is the third most frequent murine leukemia virus (MLV) integration site, driving T cell lymphomas in mice [83, 84] and rats [85, 86]. Tumors induced by retroviral insertions into the locus, that mainly clusterized around exon 1, where characterized by overexpression of the exon 1 of pvt1 and of its microRNA product, mmu-miR-1204, which is encoded by exon 1 as well. Moreover, MLV integration in the pvt1 locus variably co-occurred with insertions tagging evi5, notch1, rasgrp1, ahi1, gfi1, but not myc [84]. The mutual exclusivity between pvt1 and myc genomic rearrangements reinforces the hypothesis of their reciprocal relationship in terms of expression and biological function.
A number of genome-wide association studies on Hodgkin’s lymphoma (HL), diffuse large B cell lymphoma (DLBCL) and follicular lymphoma (FL) identified novel susceptibility loci at 8q24.21 near or in the PVT1 gene. In particular, rs2019960, encompassing intron 6 of PVT1 and rs2608053, localized telomerically to PVT1, were associated with classical HL risk, with poor correlation between each other [59]. The rs2608053 susceptibility locus (GG vs AG + AA) was also predictive of patients’ outcome in terms of progression free and overall survival in HL [60]. Two independent variants, rs13255292 and rs4733601, located telomerically to the 8q24 region, in close proximity to PVT1, were recognized as risk factors for DLBCL [61] and one of them (rs13255292) was confirmed in the Eastern Asian population [62]. Moreover, a new susceptibility locus (rs13254990) mapping near PVT1, was associated with FL risk [65]. In DLBCL, focal deletions of the PVT1 promoter was suggested to promote MYC overexpression and a double-hit-like expression pattern in germinal center type tumors lacking MYC and/or BCL2 rearrangements [63].
Despite the frequency of genomic alterations and rearrangements, few studies investigated the functional role of PVT1 in lymphomas. Recently, a novel cell line, named AMU-ML2, characterized by the occurrence of a homogeneously staining region at the 8q24 locus and containing more than 20 copies of the entire MYC and PVT1 genes, has been established from a DLBCL patient at diagnosis. Of note, AMU-ML2 cells expressed elevated levels of MYC, PVT1 and circPVT1 and were resistant to vincristine, suggesting a potential link between PVT1 and drug resistance in DLBCL [64]. This hypothesis is in line with previous reports on PVT1-mediated resistance to cisplatin in gastric and ovarian cancers [87].
Multiple myeloma
PVT1 is frequently involved in translocations occurring in multiple myeloma (MM) and murine plasmacytoma, where the t(6;15) (igκ-pvt1) and the t(15;16) (pvt1–igλ) fusion genes [88, 89] have been reported to upregulate PVT1 expression [90]. Various partners loci were described in MM with 8q24 abnormalities, including 4p16, 4q13, 13q13, 14q32, and 16q23–24 [66]. In particular, the PVT1-NBEA and PVT1-WWOX chimeras were highly expressed in the AMU-MM1 and RPMI8226 cell lines harboring t(8;13)(q24;q13) and der(16)t(16;22)ins(16;8)(q23;q24) rearrangements, respectively [66]. Moreover, co-amplification of MYC and PVT1 has been also reported in MM [67].
Elevated PVT1 levels were detected in MM BM cells compared with normal tissue [68] and primarily in MYC-rearranged MM cases [69]. In particular, high PVT1 was associated with MM carrying recurrent MYC fusion genes (with IGH, IGK, IGL, TXNDC5/BMP6, FOXO3 and FAM46C partner genes) or complex rearrangements involving more than five loci, or hyperdiploid cases, that also overexpressed MYC [69]. Despite the weak correlation observed between MYC and PVT1 expression in MM, a MYC binding site in the PVT1 gene was experimentally validated in MM cells, thus suggesting a positive feedback loop between the two genes, sustaining their elevated expression [69, 91]. Taken together, this evidence could depict a novel molecular paradigm underlying the pathogenesis of 8q24 rearrangement-positive MM. PVT1 knockdown in MM cell lines inhibited cell proliferation and promoted apoptosis [48] through restoration of expression of miR-203a [68] (Fig. 3). Indeed, PVT1 acts as a miR-203a sponge and silencing of miR-203a reversed the PVT1-knockdown phenotype. A similar function has been recently suggested for circPVT1, whose ectopic expression enhanced the proliferation rate of MM models, suppressed apoptosis and expanded the stem cell compartment [70]. Moreover, recent findings point to PVT1 and circPVT1 role in treatment response and resistance. PVT1 was downregulated by BET inhibitors, along with MYC [92]. However, it was not altered by inhibitors of MYC transcriptional activity, suggesting a BRD4-mediated co-regulation of the two genes, rather than a MYC-dependent expression of the lncRNA. Moreover, circPVT1 is overexpressed in glucocorticoid resistant cells and its downregulation enhanced sensitivity to glucocorticoid treatment, induced apoptosis and inhibited cell proliferation in resistant cell lines and xenograft models through upregulation of Caspase-3 and PARP and downregulation of BCL2 [70].
PVT1 and circPVT1: MYC partners in crime and beyond
Due to their close proximity at the 8q24 locus, PVT1 and MYC are often considered tween players, and a positive interaction feedback loop has been demonstrated in solid tumors [93] and in APL [46] and MM [91], among hematological malignancies. It is doubtless that their co-expression does not occur by chance. Indeed, normal tissues generally express MYC but very low levels of PVT1, while transformed cells also display elevated PVT1 [91, 93]. Moreover, BRD4 has been suggested to regulate the expression of both PVT1 and MYC in MM [92]. By comparing transcripts correlated with PVT1 expression across myeloid and B cell malignancies (adult and pediatric AML, pediatric ALL and DLBCL, data available through the cBioPortal database, https://www.cbioportal.org) we identified a core of 169 common genes, showing a positive or negative correlation with PVT1 (Spearman q ≤ 0.05, Additional file 1). Of note, 9 of them mapped at the 8q24 locus (CYC1; TSTA3; SLC39A4; PUF60; SHARPIN; ADCK5; MFSD3; FBXL6; HSF1, fold discovery rate [FDR] = 0.0002 from http://www.webgestalt.org/2017/option.org [94]), suggesting a positional effect.
The biological consequences of PVT1 and circPVT1 alterations are often attributed to the downstream deregulation of MYC. Evidence obtained across hematological malignancies indicates that the regulatory role exerted on cell proliferation by PVT1 (in AML, T-ALL, BL, MM) and circPVT1 (in T-ALL, B-ALL) is mediated by MYC, as also observed in a number of solid tumors [95, 96], and this regulation is active at post-transcriptional level (Fig. 3). Several studies have suggested that PVT1 enhances the stability of MYC protein by preventing its phosphorylation at threonine 58 and subsequent degradation [95, 97]. Moreover, PVT1 can also bind to MYC transcript and regulate its expression [97]. No correlation between MYC transcript and PVT1 was reported in adult and pediatric AML, pediatric ALL and DLBCL, differently from observations obtained in pan-cancer cohorts, which is indicative of a lower frequency of genomic amplification occurring at the 8q24 locus [93]. Conversely, enrichment of a MYC-related signature was identified among PVT1 coexpressed genes in hematological malignancies (QTRT1; TSFM; ZNF593; NDUFAF4; CCDC124; MON1A; RRP9; ISOC2, adjusted p = 0.04, MSigDB oncogenic signatures, https://amp.pharm.mssm.edu/Enrichr/enrich [98]), further reinforcing the post-transcriptional nature of the MYC-PVT1 interplay.
Along with MYC-related genes, pathway analysis highlighted a significant enrichment of transcripts involved in transcription, RNA metabolism, translation, oxidative phosphorylation, purine and pyrimidine metabolism (Additional file 2), suggesting a potential role of PVT1 in these cellular processes. The correlating genes may be either regulated by PVT1 and/or co-regulated with the lncRNA itself. In addition, PVT1 was reported to recruit EZH2 [99] to LATS2 [100], CDKN2B and CDKN2A [101] promoters, in order to repress their transcription, suggesting a MYC-independent activity. Although this evidence was obtained in solid tumor models, the involved genes, and in particular the epigenetic regulator EZH2 are well known also in hematological malignancies [102], thus deserving further investigation.
The oncogenic role of PVT1 and circPVT1 has also been linked to their function as competing endogenous RNA (ceRNA) through binding of tumor suppressor microRNAs and circPVT1 could serve as sponges for RNA-binding proteins (RBPs, Fig. 3). Indeed, binding sites for EIF4A3, U2AF65, AGO2, AUF1, DGCR8, FUS, HNRNPC, PTB, TAF15, TDP43, TIA1, TIAL1, LIN28A were predicted on circPVT1, with the first 3 RBPs showing the strongest evidence (https://circinteractome.nia.nih.gov/index.html). Among the microRNAs regulated by PVT1, miR-26b, miR-203a, miR-214, miR-424 and miR-497 were reported to be deregulated and play a role in the pathogenesis of lymphoma [103,104,105,106,107,108] and/or MM [68, 109,110,111,112,113] and/or leukemia [114,115,116,117,118,119,120,121]. mir-26b appeared among those showing a significant interaction with the core of PVT1-coexpressed transcripts (p ≤ 0.05, mirTarbase, https://amp.pharm.mssm.edu/Enrichr/enrich), along with miR-186 and miR-16, which are also sponged by PVT1 and are involved in the regulation of cell cycle and apoptosis [122]. Of note, it was recently reported that PVT1 also regulates cell migration and angiogenesis through miR-26b and miR-186, along with direct interaction with RBPs and/or signaling molecules. Indeed, PVT1-binding of miR-186 led to upregulation of the autophagy-promoting genes ATG7 and Beclin1, with increased migration of glioma vascular endothelial cells [123]. Similarly, PVT1 enhanced in vitro vascular tube formation of HUVECs by enforcing the expression of CTGF and ANGPT2, through miR-26b suppression [124]. The pro-angiogenic function of PVT1 is also exerted through direct interaction with phospho-STAT3, leading to protein stabilization and activation of the downstream pathway, resulting in VEGFA upregulation in gastric cancer [125]. Moreover, PVT1 regulates VEGF in non-small-cell lung cancer and ANGPTL4 in cholangiocarcinoma by acting as miR-29c sponge [126] and by binding to the epigenetic modification complex PRC2 [127], respectively. These data, obtained in solid tumors, can offer new insights in hemato-oncology, and in particular in MM, where angiogenesis is a hallmark of disease progression [128].
Recent evidence on the role of PVT1 in immune system opens a new scenario, in which the link with MYC has not been completely addressed. Indeed, MYC is a downstream target of PVT1 in G-MDSC, but its involvement in the regulation of their immune suppressive function, including ROS production and ARG1 activity, has not been fully elucidated [38]. Of note, Zheng et al. proposed a microenvironmental regulation of PVT1 expression, mediated by HIF-1α under hypoxic stress [38]. Similarly, levels of circPVT1 and other circRNAs are physiologically regulated or pathologically deregulated by RNase L in immune cells, resulting in changes of PKR activity and, consequently in T cell functionality [12]. These data point to novel putative MYC-independent functions of PVT1 and circPVT1 in (anti-tumor) immune response.
Conclusions
The scenario depicted by recent data discussed in this review suggests a double edge of PVT1 and circPVT1 in the hematopoietic system: under non-tumorigenic conditions, circPVT1 has a positive role in the regulation of protective immune responses and pathological effects in case of autoimmune reactions (Fig. 4). However, PVT1 and circPVT1 potentiate malignant cells, while hampering anti-tumor immune response during cancer progression (Fig. 4). No transforming ability of PVT1 and circPVT1 per se has been demonstrated in hematopoietic cells so far, suggesting a role in tumor progression and in support of a proliferative phenotype, rather than in cancer development. Moreover, some new insights on the role of PVT1 in drug response are emerging, including the elevated expression of both PVT1 and circPVT1 in the vincristine-resistant AMU-ML2 DLBCL line [64] and the glucocorticoid-resistant phenotype promoted by circPVT1 in MM models, that is rescued by knockdown [70]. Although some of these data need to be substantiated by scientific publications, they provide a clear path to go in hemato-oncology, where targeting of PVT1 and/or circPVT1 may a be a valuable option for combination therapies. Indeed, PVT1/circPVT1-mediated immune suppression is a challenge for those therapies aiming at restoring effective anti-tumor immune responses, including monoclonal antibodies (e.g.. αPD-1, αPD-L1, αCTLA-4) and cell-based therapies (e.g. chimeric antigen receptors [CAR]-T and CAR-cytokine induced killer cells) that are emerging as preferred candidates for combination therapies and the future of anti-cancer therapy, respectively.

Consequences of PVT1 and/or circPVT1 deregulation in the hematopoietic system
Some key questions still need to be addressed regarding the role of PVT1 and circPVT1. First, most studies did not account for the diverse PVT1 isoforms and their differential expression across human cancers. Therefore, the most abundant variants expressed in the hematopoietic tissues may have been missed and little is known on the expression and function of circPVT1 isoforms in the hematopoietic cells. Most functional studies show an intermediate phenotype, suggesting a potential compensatory role of secondary isoforms, which has not been elucidated yet. Second, similarities and differences between PVT1 and the most studied circPVT1 isoform remain vague. A similar role has been proposed in ALL and MM. However, their reciprocal regulation is still unexplored and the genomic localization of circPVT1 does not facilitate the work. Their structural differences also suggest potentially diverse mechanisms of action, with circPVT1 being a preferred candidate for extracellular localization (e.g. extracellular vesicles, cell-free RNA) and for the cross-talk between cancer cells and the immune system, that deserve future investigation. So far, 8/26 PVT1 isoforms (although not including the ones expressed at high level in the hematopoietic tissues) have been detected in exosomes from cell lines, primary tumors and/or serum of patients with active tuberculosis (http://www.noncode.org), suggesting a functional role on the tumor microenvironment, especially under inflammatory conditions, that in turn favour tumor progression. Therefore, extracellular PVT1 and especially circPVT1 may have a putative role in hematological malignancies. In MM, extracellular vesicles have a supportive role during metastatization by promoting angiogenesis, uptake at distant premetastatic niches and activation of osteolytic activity [129]. Given their known role in angiogenesis, as proved in solid tumors, and their ability to shape cellular function, as demonstrated in immune cells, we can hypothesize that PVT1 and circPVT1 may be loaded as RNA cargo in extracellular vesicles released by plasma cells, in order to instruct the tumor microenvironment (e.g. fibroblasts) and promote bone metastasis.
A better understanding of all these topics will help to define targeted therapeutic interventions acting on the good and the bad of the hematopoietic system, with the aim of weakening the malignant cells and reactivate the anti-tumor immune response.
Availability of data and materials
The datasets analysed during the current study are available in the [BioPortal for cancer genomics repository, [http://www.cbioportal.org/].
Abbreviations
- AEL:
-
Acute erythroleukemia
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- AMPL-amp:
-
AML cases carrying MYC amplifications as dmin, hsr or ring chromosomes
- APL:
-
Acute promyelocytic leukemia
- ATRA:
-
All-trans retinoic acid
- BET:
-
Bromodomain and extra-terminal domain
- BL:
-
Burkitt lymphoma
- BM:
-
Bone marrow
- CAR:
-
Chimeric antigen receptor
- ceRNA:
-
Competing endogenous RNA
- circRNA:
-
Circular RNA
- CLL:
-
Chronic lymphocytic leukemia
- DLBCL:
-
Diffuse large B cell lymphoma
- dmin:
-
Double minutes chromosomes
- ds:
-
Double stranded
- FL:
-
Follicular lymphoma
- HL:
-
Hodgkin’s lymphoma
- hsr:
-
Homogeneously staining region
- Ig:
-
Immunoglobulin
- G-MDSC:
-
Granulocytic myeloid-derived suppressor cells
- IFN:
-
Interferon
- lncRNA:
-
Long ncRNA
- MLV:
-
Murine leukemia virus
- MM:
-
Multiple myeloma
- ncRNA:
-
Non coding RNA
- ORF:
-
Open reading frame
- PB:
-
Peripheral blood
- PVT1 :
-
Human Plasmacytoma Variant Translocation 1
- ROS:
-
Reactive oxygen species
- SLE:
-
Systemic lupus erythematosus
- Treg:
-
Regulatory T cells
References
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004.
Calin GA, Gong LC, Ferracin M, Hyslop T, Spizzo R, Sevignani C, et al. Ultraconserved regions encoding ncRNAs are altered in human Leukemias and carcinomas. Cancer Cell. 2007;12:215–29 Cell Press.
Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T. Emerging roles of long non-coding RNA in cancer. Cancer Sci, Blackwell Publishing Ltd. 2018;109:2093–100.
Vannini I, Fanini F, Fabbri M. Emerging roles of microRNAs in cancer. Curr Opin Genet Dev, Elsevier Ltd. 2018;48:128–33.
Spizzo R, Almeida MI, Colombatti A, Calin GA. Long non-coding RNAs and cancer: A new frontier of translational research. Oncogene. 2012;31:4577–87.
Ulitsky I, Bartel DP. XLincRNAs: Genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46 Cell Press.
Mercer TR, Mattick JS. Understanding the regulatory and transcriptional complexity of the genome through structure. Genome Res. 2013;23:1081–8.
Vannini I, Wise PM, Challagundla KB, Plousiou M, Raffini M, Bandini E, et al. Publisher correction: transcribed ultraconserved region 339 promotes carcinogenesis by modulating tumor suppressor microRNAs. Nat Commun. 2018;9:160.
Ebbesen KK, Hansen TB, Kjems J. Insights into circular RNA biology. RNA Biol. 2017; 14(8):1035–45 Taylor and Francis Inc.
Li X, Yang L, Chen LL. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol Cell. 2018;71:428–42 Cell Press.
Kristensen LS, Okholm TLH, Venø MT, Kjems J. Circular RNAs are abundantly expressed and upregulated during human epidermal stem cell differentiation. RNA Biol. 2018;15:280–91 Taylor and Francis Inc.
Liu CX, Li X, Nan F, Jiang S, Gao X, Guo SK, et al. Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell. 2019;177:865–880.e21 Cell Press.
Dudekula DB, Panda AC, Grammatikakis I, De S, Abdelmohsen K, Gorospe M. Circinteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016;13:34–42 Taylor and Francis Inc.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7(2).
Panda AC, Grammatikakis I, Kim KM, De S, Martindale JL, Munk R, et al. Identification of senescence-associated circular RNAs (SAC-RNAs) reveals senescence suppressor CircPVT1. Nucleic Acids Res. 2017;45:4021–35 Oxford University Press.
Fang S, Zhang L, Guo J, Niu Y, Wu Y, Li H, et al. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018;46:D308–14 Oxford University Press.
Salehi M, Sharifi M, Bagheri M. Knockdown of Long Noncoding RNA Plasmacytoma Variant Translocation 1 with Antisense Locked Nucleic Acid GapmeRs Exerts Tumor-Suppressive Functions in Human Acute Erythroleukemia Cells Through Downregulation of C-MYC Expression. Cancer Biother Radiopharm. 2019;34:371–9 Mary Ann Liebert Inc.
Schwarzer A, Emmrich S, Schmidt F, Beck D, Ng M, Reimer C, et al. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat Commun Group. 2017;8(1):218 Nature Publishing.
Verduci L, Ferraiuolo M, Sacconi A, Ganci F, Vitale J, Colombo T, et al. The oncogenic role of circPVT1 in head and neck squamous cell carcinoma is mediated through the mutant p53/YAP/TEAD transcription-competent complex. Genome Biol. 2017;18(1):237 BioMed Central Ltd.
Chen J, Li Y, Zheng Q, Bao C, He J, Chen B, et al. Circular RNA profile identifies circPVT1 as a proliferative factor and prognostic marker in gastric cancer. Cancer Lett. 2017;388:208–19 Elsevier Ireland Ltd.
Panda AC, De S, Grammatikakis I, Munk R, Yang X, Piao Y, et al. High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res. 2017;45(12):e116 Oxford University Press.
Tashiro K, Tseng Y-Y, Konety B, Bagchi A. Role of non-coding RNA PVT1 in regulating MYC in human cancer. J Urol. 2017;194.
Derderian C, Orunmuyi AT, Oluwabunmi Olapade-Olaopa E, Ogunwobi OO. PVT1 signaling is a mediator of cancer progression. Front Oncol. 2019;9 Frontiers Media S.A.
Liu L, Wang J, Khanabdali R, Kalionis B, Tai X, Xia S. Circular RNAs: Isolation, characterization and their potential role in diseases. RNA Biol. 2017;14:1715–21 Taylor and Francis Inc.
Chen YG, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol. 2017;18:962–72.
Sigdel KR, Cheng A, Wang Y, Duan L, Zhang Y. The emerging functions of long noncoding RNA in immune cells: autoimmune diseases. J Immunol Res. 2015;2015:848790.
Gillinder KR, Tuckey H, Bell CC, Magor GW, Huang S, Ilsley MD, et al. Direct targets of pSTAT5 signalling in erythropoiesis. PLoS One. 2017;12:e0180922.
Aryankalayil MJ, Chopra S, Levin J, Eke I, Makinde A, Das S, et al. Radiation-Induced Long Noncoding RNAs in a Mouse Model after Whole-Body Irradiation. Radiat Res. 2018;189:251 Radiation Research Society.
Barsotti AM, Beckerman R, Laptenko O, Huppi K, Caplen NJ, Prives C. p53-dependent induction of PVT1 and miR-1204. J Biol Chem. 2012;287:2509–19.
Bronson PG, Chang D, Bhangale T, Seldin MF, Ortmann W, Ferreira RC, et al. Common variants at PVT1, ATG13-AMBRA1, AHI1 and CLEC16A are associated with selective IgA deficiency. Nat Genet. 2016;48:1425–9.
Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci U S A. 2004;101:1981–6.
Cong Y, Feng T, Fujihashi K, Schoeb TR, Elson CO. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A. 2009;106:19256–61.
Ludvigsson JF, Neovius M, Hammarström L. Association between IgA deficiency & other autoimmune conditions: a population-based matched cohort study. J Clin Immunol. 2014;34:444–51.
Huang H, Zeqiraj E, Dong B, Jha BK, Duffy NM, Orlicky S, et al. Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Mol Cell. 2014;53:221–34.
Eftekharian MM, Ghafouri-Fard S, Soudyab M, Omrani MD, Rahimi M, Sayad A, et al. Expression analysis of long non-coding RNAs in the blood of multiple sclerosis patients. J Mol Neurosci. 2017;63:333–41 Springer New York LLC.
Yu X, Zhe Z, Tang B, Li S, Tang L, Wu Y, et al. α-Asarone suppresses the proliferation and migration of ASMCs through targeting the lncRNA-PVT1/miR-203a/E2F3 signal pathway in RSV-infected rats. Acta Biochim Biophys Sin (Shanghai). 2017;49:598–608.
Huang W, Lan X, Li X, Wang D, Sun Y, Wang Q, et al. Long non-coding RNA PVT1 promote LPS-induced septic acute kidney injury by regulating TNFα and JNK/NF-κB pathways in HK-2 cells. Int Immunopharmacol. 2017;47:134–40.
Zheng Y, Tian X, Wang T, Xia X, Cao F, Tian J, et al. Long noncoding RNA Pvt1 regulates the immunosuppression activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Mol Cancer. 2019;18(1):61 BioMed Central Ltd.
Asker C, Mareni C, Coviello D, Ingvarsson S, Sessarego M, Origone P, et al. Amplification of c-myc and pvt-1 homologous sequences in acute nonlymphatic leukemia. Leuk Res. 1988;12:523–7.
Storlazzi CT, Fioretos T, Paulsson K, Strömbeck B, Lassen C, Ahlgren T, et al. Identification of a commonly amplified 4.3 Mb region with overexpression of C8FW, but not MYC in MYC-containing double minutes in myeloid malignancies. Hum Mol Genet. 2004;13:1479–85.
L’Abbate A, Tolomeo D, Cifola I, Severgnini M, Turchiano A, Augello B, et al. MYC-containing amplicons in acute myeloid leukemia: genomic structures, evolution, and transcriptional consequences. Leukemia. 2018:32(10):2304.
Chinen Y, Sakamoto N, Nagoshi H, Taki T, Maegawa S, Tatekawa S, et al. 8q24 amplified segments involve novel fusion genes between NSMCE2 and long noncoding RNAs in acute myelogenous leukemia. J Hematol Oncol. 2014;7:68.
El-Khazragy N, Elayat W, Matbouly S, Seliman S, Sami A, Safwat G, et al. The prognostic significance of the long non-coding RNAs “CCAT1, PVT1” in t (8;21) associated acute myeloid leukemia. Gene. 2019;707:172–77.
Delás MJ, Sabin LR, Dolzhenko E, Knott SR, Munera Maravilla E, Jackson BT, et al. lncRNA requirements for mouse acute myeloid leukemia and normal differentiation. Elife. 2017;6.
Izadifard M, Pashaiefar H, Yaghmaie M, Montazeri M, Sadraie M, Momeny M, et al. Expression analysis of PVT1, CCDC26, and CCAT1 long noncoding RNAs in acute myeloid leukemia patients. Genet Test Mol Biomarkers. 2018;22:593–8 Mary Ann Liebert Inc.
Zeng C, Yu X, Lai J, Yang L, Chen S, Li Y. Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J Hematol Oncol. 2015;8:126 BioMed Central Ltd.
Gómez-Seguí I, Sánchez-Izquierdo D, Barragán E, Such E, Luna I, López-Pavía M, et al. Single-nucleotide polymorphism array-based karyotyping of acute promyelocytic leukemia. PLoS One. 2014;9(6). Public Library of Science.
Houshmand M, Yazdi N, Kazemi A, Atashi A, Hamidieh AA, Anjam Najemdini A, et al. Long non-coding RNA PVT1 as a novel candidate for targeted therapy in hematologic malignancies. Int J Biochem Cell Biol. 2018;98:54–64 Elsevier Ltd.
Salehi M, Sharifi M. Induction of apoptosis and necrosis in human acute erythroleukemia cells by inhibition of 47Tlong non-coding RNA PVT1. Mol Biol Res Commun. 2018;7(2):89–96.
Hu J, Han Q, Gu Y, Ma J, McGrath M, Qiao F, et al. Circular RNA PVT1 expression and its roles in acute lymphoblastic leukemia. Epigenomics. 2018;10:723–32 Future Medicine Ltd.
Gaffo E, Boldrin E, Dal Molin A, Bresolin S, Bonizzato A, Trentin L, et al. Circular RNA differential expression in blood cell populations and exploration of circRNA deregulation in pediatric acute lymphoblastic leukemia. Sci Rep. 2019;9(1):14670.
Yazdi N, Houshmand M, Atashi A, Kazemi A, Najmedini AA, Zarif MN. Long noncoding RNA PVT1: potential oncogene in the development of acute lymphoblastic leukemia. Turkish J Biol. 2018;42:405–13 Scientific and Technical Research Council of Turkey.
Macchia G, Lonoce A, Venuto S, Macrí E, Palumbo O, Carella M, et al. A rare but recurrent t (8;13)(q24;q14) translocation in B-cell chronic lymphocytic leukaemia causing MYC up-regulation and concomitant loss of PVT1, miR-15/16 and DLEU7. Br J Haematol. 2016;172:296–9 Blackwell Publishing Ltd.
Sun LK, Showe LC, Croce CM. Analysis of the 3′ flanking region of the human c-myc gene in lymphomas with the t (8;22) and t (2;8) chromosomal translocations. Nucleic Acids Res. 1986;14:4037–50.
Henglein B, Synovzik H, Groitl P, Bornkamm GW, Hartl P, Lipp M. Three breakpoints of variant t (2;8) translocations in Burkitt’s lymphoma cells fall within a region 140 kilobases distal from c-myc. Mol Cell Biol. 1989;9:2105–13 American Society for Microbiology.
Rack KA, Delabesse E, Radford-Weiss I, Bourquelot P, Le Guyader G, Vekemans M, et al. Simultaneous detection of MYC, BVR1, and PVT1 translocations in lymphoid malignancies by fluorescence in situ hybridization. Genes Chromosomes Cancer. 1998;23:220–6.
Grande BM, Gerhard DS, Griner NB, Casper C, Namirembe C, Omoding A, et al. Burkitt lymphoma genome sequencing project (BLGSP): integrative genomic and Transcriptomic characterization of Burkitt lymphoma. Blood. 2017;130:39.
Zheng C, Xiao Y, Li Y, He D. Knockdown of long non-coding RNA PVT1 inhibits the proliferation of raji cells through cell cycle regulation. Oncol Lett. 2019;18:1225–34 Spandidos Publications.
Enciso-Mora V, Broderick P, Ma Y, Jarrett RF, Hjalgrim H, Hemminki K, et al. A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat Genet. 2010;42:1126–30.
Ghesquières H, Larrabee BR, Casasnovas O, Maurer MJ, McKay JD, Ansell SM, et al. A susceptibility locus for classical Hodgkin lymphoma at 8q24 near MYC/PVT1 predicts patient outcome in two independent cohorts. Br J Haematol. 2018;180:286–90. Blackwell Publishing Ltd.
Cerhan JR, Berndt SI, Vijai J, Ghesquières H, McKay J, Wang SS, et al. Genome-wide association study identifies multiple susceptibility loci for diffuse large B cell lymphoma. Nat Genet. 2014;46:1233–8 Nature Publishing Group.
Bassig BA, Cerhan JR, Au WY, Kim HN, Sangrajrang S, Hu W, et al. Genetic susceptibility to diffuse large B-cell lymphoma in a pooled study of three eastern Asian populations. Eur J Haematol. 2015;95:442–8 Blackwell Publishing Ltd.
Hilton LK, Tang J, Ben-Neriah S, Alcaide M, Jiang A, Grande BM, et al. The double-hit signature identifies double-hit diffuse large B-cell lymphoma with genetic events cryptic to FISH. Blood. 2019;134:1528–32 American Society of Hematology.
Mizuno S, Hanamura I, Ota A, Karnan S, Kanasugi J, Nakamura A, et al. Establishment and characterization of a novel vincristine-resistant diffuse large B-cell lymphoma cell line containing the 8q24 homogeneously staining region. FEBS Open Bio. 2018;8:1977–91 Wiley Blackwell.
Skibola CF, Berndt SI, Vijai J, Conde L, Wang Z, Yeager M, et al. Genome-wide association study identifies five susceptibility loci for follicular lymphoma outside the HLA region. Am J Hum Genet. 2014;95:462–71 Cell Press.
Nagoshi H, Taki T, Hanamura I, Nitta M, Otsuki T, Nishida K, et al. Frequent PVT1 rearrangement and novel chimeric genes PVT1-NBEA and PVT1-WWOX occur in multiple myeloma with 8q24 abnormality. Cancer Res. 2012;72:4954–62.
Bakkus MH, Brakel-van Peer KM, Michiels JJ, van’t Veer MB, Benner R. Amplification of the c-myc and the pvt-like region in human multiple myeloma. Oncogene. 1990;5:1359–64.
Yang M, Zhang L, Wang X, Zhou Y, Wu S. Down-regulation of miR-203a by lncRNA PVT1 in multiple myeloma promotes cell proliferation. Arch Med Sci. 2018;14:1333–9 Termedia Publishing House Ltd.
Mikulasova A, Ashby C, Tytarenko RG, Qu P, Rosenthal A, Dent JA, et al. Microhomology-mediated end joining drives complex rearrangements and over expression of MYC and PVT1 in multiple myeloma. Haematologica. 2019; haematol.2019.217927. Ferrata Storti Foundation (Haematologica).
Wan X-Y, Chu Z-B, Hu Y, Sun C-Y, Zou J. CircPVT1 inhibit apoptosis and enhance drug resistance in multiple myeloma. Blood. 2017;130:3085.
Akagi K. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32:523D–5527. Oxford University Press (OUP).
Nakamura Y, Kayano H, Kakegawa E, Miyazaki H, Nagai T, Uchida Y, et al. Identification of SUPT3H as a novel 8q24/MYC partner in blastic plasmacytoid dendritic cell neoplasm with t (6;8)(p21;q24) translocation. Blood Cancer J. 2015;5:e301.
He RQ, Qin MJ, Lin P, Luo YH, Ma J, Yang H, et al. Prognostic significance of LncRNA PVT1 and its potential target gene network in human cancers: a comprehensive inquiry based upon 21 Cancer types and 9972 cases. Cell Physiol Biochem. 2018;46:591–608 S Karger AG.
Ma X, Jin W, Zhao M, Zhang W, Li J, Wang K. Long noncoding RNA profiling reveals an abundant Crnde that inhibits granulocytic differentiation in APL. Blood. 2017;130:3799.
Fan H, Zhu JH, Yao XQ. Long non-coding RNA PVT1 as a novel potential biomarker for predicting the prognosis of colorectal cancer. Int J Biol Markers. 2018;33:415–22 SAGE Publications Ltd.
Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–8.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–8.
Brown JR, Hanna M, Tesar B, Werner L, Pochet N, Asara JM, et al. Integrative genomic analysis implicates gain of PIK3CA at 3q26 and MYC at 8q24 in chronic lymphocytic leukemia. Clin Cancer Res. 2012;18:3791–802.
Li Y, Hu S, Wang SA, Li S, Huh YO, Tang Z, et al. The clinical significance of 8q24/MYC rearrangement in chronic lymphocytic leukemia. Mod Pathol. 2016;29:444–51 Nature Publishing Group.
Rinaldi A, Mian M, Kwee I, Rossi D, Deambrogi C, Mensah AA, et al. Genome-wide DNA profiling better defines the prognosis of chronic lymphocytic leukaemia. Br J Haematol. 2011;154:590–9.
Houldsworth J, Guttapalli A, Thodima V, Yan XJ, Mendiratta G, Zielonka T, et al. Genomic imbalance defines three prognostic groups for risk stratification of patients with chronic lymphocytic leukemia. Leuk Lymphoma. 2014;55:920–8 Informa Healthcare.
Chapiro E, Lesty C, Gabillaud C, Durot E, Bouzy S, Armand M, et al. “Double-hit” chronic lymphocytic leukemia: An aggressive subgroup with 17p deletion and 8q24 gain. Am J Hematol. 2018;93:375–82 Wiley-Liss Inc.
Graham M, Adams JM, Cory S. Murine T lymphomas with retroviral inserts in the chromosomal 15 locus for plasmacytoma variant translocations. Nature. 1985;314:740–3.
Beck-Engeser GB, Lum AM, Huppi K, Caplen NJ, Wang BB, Wabl M. Pvt1-encoded microRNAs in oncogenesis. Retrovirology. 2008:5;4.
Lemay G, Jolicoeur P. Rearrangement of a DNA sequence homologous to a cell virus junction fragment in several Moloney murine leukemia virus induced rat thymomas. Proc Natl Acad Sci U S A. 1984;81:38–42.
Villeneuve L, Rassart E, Jolicoeur P, Graham M, Adams JM. Proviral integration site Mis-1 in rat thymomas corresponds to the pvt-1 translocation breakpoint in murine plasmacytomas. Mol Cell Biol. 1986;6:1834–7 American Society for Microbiology.
Liu E, Liu Z, Zhou Y, Mi R, Wang D. Overexpression of long non-coding RNA PVT1 in ovarian cancer cells promotes cisplatin resistance by regulating apoptotic pathways. Int J Clin Exp Med. 2015;8:20565–72 E-Century Publishing Corporation.
Huppi K, Siwarski D. Chimeric transcripts with an open reading frame are generated as a result of translocation to the Pvt-1 region in mouse B-cell tumors. Int J Cancer. 1994;59:848–51.
McNeil N, Joong SK, Ried T, Janz S. Extraosseous IL-6 transgenic mouse plasmacytoma sometimes lacks Myc-activating chromosomal translocation. Genes Chromosom Cancer. 2005;43(2):137–46.
Huppi K, Siwarski D, Skurla R, Klinman D, Mushinski JF. Pvt-1 transcripts are found in normal tissues and are altered by reciprocal (6;15) translocations in mouse plasmacytomas. Proc Natl Acad Sci U S A. 1990;87:6964–8.
Carramusa L, Contino F, Ferro A, Minafra L, Perconti G, Giallongo A, et al. The PVT-1 oncogene is a Myc protein target that is overexpressed in transformed cells. J Cell Physiol. 2007;213:511–8.
Homma K, Oda T, Murakami YG, Watanabe S, Ishihara R, Kuroda Y, et al. Long noncoding RNA PVT1 and MYC are co-regulated by Bromodomain protein BRD4 in multiple myeloma and associated with disease Progressionn. Blood. 2017;130:4397.
Colombo T, Farina L, Macino G, Paci P. PVT1: a rising star among oncogenic long noncoding RNAs. Biomed Res Int. 2015. Hindawi Publishing Corporation.
Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–7 Oxford University Press.
Jin K, Wang S, Zhang Y, Xia M, Mo Y, Li X, et al. Long non-coding RNA PVT1 interacts with MYC and its downstream molecules to synergistically promote tumorigenesis. Cell Mol Life Sci. 2019;76:4275–89 Birkhauser Verlag AG.
Adhikary J, Chakraborty S, Dalal S, Basu S, Dey A, Ghosh A. Circular PVT1: an oncogenic non-coding RNA with emerging clinical importance. J Clin Pathol. 2019;72(8):513–9.
Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, et al. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–6 Nature Publishing Group.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–7.
Zhou Q, Chen J, Feng J, Wang J. Long noncoding RNA PVT1 modulates thyroid cancer cell proliferation by recruiting EZH2 and regulating thyroid-stimulating hormone receptor (TSHR). Tumor Biol. 2016;37:3105–13 Springer Netherlands.
Wan L, Sun M, Liu GJ, Wei CC, Zhang EB, Kong R, et al. Long noncoding RNA PVT1 promotes non-small cell lung cancer cell proliferation through epigenetically regulating LATS2 expression. Mol Cancer Ther. 2016;15(5):1082–94.
Kong R, Zhang EB, Yin DD, You LH, Xu TP, Chen WM, et al. Long noncoding RNA PVT1 indicates a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically regulating p15 and p16. Mol Cancer. 2015;14:82. BioMed Central Ltd.
Nakagawa M, Kitabayashi I. Oncogenic roles of enhancer of zeste homolog 1/2 in hematological malignancies. Cancer Sci, Blackwell Publishing Ltd. 2018;109:2342–8.
Peveling-Oberhag J, Crisman G, Schmidt A, Döring C, Lucioni M, Arcaini L, et al. Dysregulation of global microRNA expression in splenic marginal zone lymphoma and influence of chronic hepatitis C virus infection. Leukemia. 2012;26(7):1654–62.
Kasama Y, Mizukami T, Kusunoki H, Peveling-Oberhag J, Nishito Y, Ozawa M, et al. B-cell-intrinsic hepatitis C virus expression leads to B-cell- lymphomagenesis and induction of NF-κB signalling. PLoS One. 2014;9(3).
Lim EL, Trinh DL, Scott DW, Chu A, Krzywinski M, Zhao Y, et al. Comprehensive miRNA sequence analysis reveals survival differences in diffuse large B-cell lymphoma patients. Genome Biol. 2015;16:18.
Arribas AJ, Gómez-Abad C, Sánchez-Beato M, Martinez N, Dilisio L, Casado F, et al. Splenic marginal zone lymphoma: comprehensive analysis of gene expression and miRNA profiling. Mod Pathol. 2013:26(7):889–901.
Zhu Q, Li Y, Guo Y, Hu L, Xiao Z, Liu X, et al. Long non-coding RNA SNHG16 promotes proliferation and inhibits apoptosis of diffuse large B-cell lymphoma cells by targeting miR-497-5p/PIM1 axis. J Cell Mol Med. 2019;23(11):7395–405.
Troppan K, Wenzl K, Pichler M, Pursche B, Schwarzenbacher D, Feichtinger J, et al. miR-199a and miR-497 are associated with better overall survival due to increased chemosensitivity in diffuse large b-cell lymphoma patients. Int J Mol Sci. 2015;16(8):18077–95.
Jia CM, Tian YY, Quan LN, Jiang L, Liu AC. miR-26b-5p suppresses proliferation and promotes apoptosis in multiple myeloma cells by targeting JAG1. Pathol Res Pract. 2018;214(9):1388–94.
Fan F, Deng R, Qiu L, Wen Q, Zeng Y, Gao L, et al. miR-203a-3p.1 is involved in the regulation of osteogenic differentiation by directly targeting Smad9 in MM-MSCs. Oncol Lett. 2019;18(6):6339–46.
Misiewicz-Krzeminska I, Sarasquete ME, Quwaider D, Krzeminski P, Ticona FV, Paíno T, et al. Restoration of microRNA-214 expression reduces growth of myeloma cells through positive regulation of P53 and inhibition of DNA replication. Haematologica. 2013;98:640–8.
Tian F, Zhan Y, Zhu W, Li J, Tang M, Chen X, et al. MicroRNA-497 inhibits multiple myeloma growth and increases susceptibility to bortezomib by targeting Bcl-2. Int J Mol Med. 2019;43(2):1058–66.
Yu T, Zhang X, Zhang L, Wang Y, Pan H, Xu Z, et al. MicroRNA-497 suppresses cell proliferation and induces apoptosis through targeting PBX3 in human multiple myeloma. Am J Cancer Res. 2016;6(12):2880–9.
Yuan T, Yang Y, Chen J, Li W, Li W, Zhang Q, et al. Regulation of PI3K signaling in T-cell acute lymphoblastic leukemia: a novel PTEN/Ikaros/miR-26b mechanism reveals a critical targetable role for PIK3CD. Leukemia. 2017;31(11):2355–64.
He Z, Liao Z, Chen S, Li B, Yu Z, Luo G, et al. Downregulated miR-17, miR-29c, miR-92a and miR-214 may be related to BCL11B overexpression in T cell acute lymphoblastic leukemia. Asia Pac J Clin Oncol. 2018;14(5):e259–e265.
Fan FY, Deng R, Yi H, Sun HP, Zeng Y, He GC, et al. The inhibitory effect of MEG3/miR-214/AIFM2 axis on the growth of T-cell lymphoblastic lymphoma. Int J Oncol. 2017;51(1):316–26.
Zou ZJ, Fan L, Wang L, Xu J, Zhang R, Tian T, et al. miR-26a and miR-214 down-regulate expression of the PTEN gene in chronic lymphocytic leukemia, but not PTEN mutation or promoter methylation. Oncotarget. 2015;6(2):1276–85.
Pallasch CP, Patz M, Yoon JP, Hagist S, Eggle D, Claus R, et al. miRNA deregulation by epigenetic silencing disrupts suppression of the oncogene PLAG1 in chronic lymphocytic leukemia. Blood. 2009;114(15):3255–64.
Forrest ARR, Kanamori-Katayama M, Tomaru Y, Lassmann T, Ninomiya N, Takahashi Y, et al. Induction of microRNAs, mir-155, mir-222, mir-424 and mir-503, promotes monocytic differentiation through combinatorial regulation. Leukemia. 2010;24(2):460–6.
Maura F, Cutrona G, Mosca L, Matis S, Lionetti M, Fabris S, et al. Association between gene and miRNA expression profiles and stereotyped subset #4 B-cell receptor in chronic lymphocytic leukemia. Leuk Lymphoma. 2015;56(11):3150–8.
Lionetti M, Musto P, Di MMT, Fabris S, Agnelli L, Todoerti K, et al. Biological and clinical relevance of miRNA expression signatures in primary plasma cell leukemia. Clin Cancer Res. 2013;19(12):3130–42.
Wang W, Zhou R, Wu Y, Liu Y, Su W, Xiong W, et al. PVT1 promotes cancer progression via MicroRNAs. Front Oncol. 2019:9;609. Frontiers Media SA.
Ma Y, Wang P, Xue Y, Qu C, Zheng J, Liu X, et al. PVT1 affects growth of glioma microvascular endothelial cells by negatively regulating miR-186. Tumor Biol. 2017;39(3). SAGE Publications Ltd.
Zheng J, Hu L, Cheng J, Xu J, Zhong Z, Yang Y, et al. LncRNA PVT1 promotes the angiogenesis of vascular endothelial cell by targeting miR-26b to activate CTGF/ANGPT2. Int J Mol Med. 2018;42:489–96 Spandidos Publications.
Zhao J, Du P, Cui P, Qin Y, Hu C, Wu J, et al. LncRNA PVT1 promotes angiogenesis via activating the STAT3/VEGFA axis in gastric cancer. Oncogene. 2018;37:4094–4109. Nature Publishing Group.
Mao Z, Xu B, He L, Zhang G. PVT1 promotes angiogenesis by regulating miR-29c/vascular endothelial growth factor (VEGF) signaling pathway in non-small-cell lung cancer (NSCLC). Med Sci Monit. 2019;25:5418–25 International Scientific Information, Inc.
Yu Y, Zhang M, Liu J, Xu B, Yang J, Wang N, et al. Long non-coding RNA PVT1 promotes cell proliferation and migration by silencing ANGPTL4 expression in cholangiocarcinoma. Mol Ther - Nucleic Acids. 2018;13:503–13 Cell Press.
Ribatti D, Nico B, Vacca A. Multiple myeloma as a model for the role of bone marrow niches in the control of angiogenesis. Int Rev Cell Mol Biol. 2015;314:259–82 Elsevier Inc.
Colombo M, Giannandrea D, Lesma E, Basile A, Chiaramonte R. Extracellular vesicles enhance multiple myeloma metastatic dissemination. Int J Mol Sci. 2019;20(13). MDPI AG.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MG and GS conception of the work; MG, IV, GS: design of the work; MG, IV, GS: preparation of manuscript; IV: preparation of Fig. 1 and correspondence; MG, IV, GS: preparation of Fig. 3; MG: preparation of Table 1, GS: data analysis and preparation of Figs. 2 and 4, Additional files 1 and 2; CTS, GM and GS: critical discussion; CTS, GM: have contributed to drafting the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
GM reports personal fees from Amgen, personal fees from Incyte, Pfizer, Celgene, Janssen, Jazz Pharmaceuticals, Abbvie, Novartis, Daiichi Sankyo, Amgen outside the submitted work. The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Core of PVT1 coexpressed genes in adult and pediatric AML, pediatric ALL and DLBCL.
Additional file 2: Table S2.
Pathway enrichment analysis of PVT1 coexpressed genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ghetti, M., Vannini, I., Storlazzi, C.T. et al. Linear and circular PVT1 in hematological malignancies and immune response: two faces of the same coin. Mol Cancer 19, 69 (2020). https://doi.org/10.1186/s12943-020-01187-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-01187-5