
Abstract
Background
The group B Streptococcus (GBS) is a human commensal bacterium, which is capable of causing several infectious diseases in infants, and people with chronic diseases. GBS has been the most common cause of infections in urinary tract of the elders, but relatively few studies reported the urine-isolated GBS and their antimicrobial susceptibilities. Hence, we decided to investigate GBS specially isolated from urine in Suzhou, China.
Methods
27 GBS samples were isolated from urine in Suzhou, China. The PCR and agarose gel electrophoresis were used to identify the serotype distribution. Susceptibility tests were based on MIC test and Kirby–Bauer test. Genome were sequenced via Illumina Hiseq platform and assembled by SPAdes. Genomes of five isolates were sequenced and submitted to NCBI genome database. The sequencing files in fastq format were submitted to NCBI SRA database.
Results
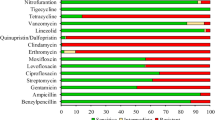
Five serotypes were identified. The resistant rates measured for tetracycline, erythromycin, clindamycin and fluoroquinolones were 74.1, 63.0, 44.4 and 48.1%, respectively. 18.5% of the isolates were nonsusceptible to nitrofurantoin. The resistance to tetracycline was mainly associated with the gene tetM. The erythromycin resistance was mainly associated with the genes ermB and mefE. The genes ermB and lnuB were the prevalent genes in cMLSB type. No known nitrofurantoin resistance gene was found in nitrofurantoin-nonsusceptible GBS.
Conclusions
Five serotypes were identified in our study. High rates of GBS isolates were resistant to tetracycline, erythromycin, clindamycin and fluoroquinolones. The genes ermB and lnuB occupied high rates in cMLSB phenotype.
Similar content being viewed by others


Background
Group B Streptococcus (GBS, i.e. Streptococcus agalactiae) is a gram-positive microbe with bacterial morphology of sphere and oval observed via microscope [1]. Tilapia mossambica and cow are susceptible to GBS, leading to the reduced milk production [2, 3]. GBS can be divided into ten serotypes, Ia, Ib, and II–IX, based on variant capsular polysaccharide. The serotypes Ia, Ib, III and V are the predominant pathogens, primarily liable for human infection [4]. GBS has traditionally been considered as a perinatal pathogen, in relation to certain infectious diseases in pregnant or postpartum women encomapassing endometritis, septicemia, chorioamnionitis and urinary tract infection [5]. Furthermore, GBS has been proved causing severe diseases in infants, such as pneumonia and meningitis [6]. Infectious pattern in infants were separated into two ways: mother-to-child vertically transmitted infection and birth canal transmission [7]. Screening for GBS in pregnant women via swabs taken from the vagina, of which the colonized GBS were mostly harmless, following by antibiotic treatment to prevent the transmission to the babies.
GBS can also cause asymptomatic bacteriuria, cystitis, pyelonephritis, urethritis and urosepsis in urinary tract [8]. In a series of studies, bacteremia and urinary tract infections caused by GBS were the most common diagnoses among people over the age of 70, with 13 of 33 cases (39.4%) [9]. Previous studies indicated that all serotypes of GBS are susceptible to penicillin and have high rates of resistance to tetracycline, clindamycin and erythromycin [10], but little attention was paid to molecular mechanisms underlying those antimicrobial susceptibilities of GBS in urinary tract. A total of 27 GBS samples were isolated from urine in this study to investigate the serotype distribution and antibiotic susceptibilities in Suzhou, China.
Methods
Samples collecting and typing
All the GBS samples were isolated from urine between November 2015 and March 2017 in Suzhou Hospital of Traditional Chinese Medicine, Suzhou, China. The isolates were cultured on Blood agar medium at 37 °C with 5% CO2 for 24 h. The DNA was extracted via the Ezup Column Bacteria Genomic DNA Purification Kit. The polymerase chain reaction (PCR) and agarose gel electrophoresis were applied (the primer sequences are in Additional file 1: Table S1, the PCR amplification system is in Additional file 1: Table S2 and the procedure of PCR is in Additional file 1: Table S3) to determine the serotype according to the expectation of electrophoresis result (Additional file 1: Table S4).
Antimicrobial susceptibility test and drug resistance gene PCR
Susceptibility tests to tetracycline, erythromycin, clindamycin, fluoroquinolones, nitrofurantoin, vancomycin, linezolid, quinupristin–dalfopristin and tigecycline were based on the MIC test. Susceptibility to ceftriaxone and penicillin was based on the Kirby–Bauer test recommended by the clinical and laboratory standards institute (CLSI) guidelines [11]. The clindamycin-induced resistance was detected based on D test [12]. The PCR of drug resistance genes was applied to find 12 resistance genes for tetracycline, erythromycin and clindamycin (Additional file 1: Table S5).
Genome sequencing and bioinformatics analysis
Genomes of four isolates (No.8, No.11, No.17 and No.24) showing multiple resistances and one isolate (No.20) indicating susceptive to all antibiotic were sequenced. The DNA was extracted by QIAGEN DNeasy® Blood & Tissue and sequenced via Illumina Hiseq platform. The genomes were assembled by SPAdes [13] and the k-mer value was set to 127. The scaffolds < 1 kb in length were dropped. The genome sequences were submitted to Antibiotic Resistance Genes Database (ARDB, http://ardb.cbcb.umd.edu/) [14] to query drug resistance genes. The genes were predicted by Prokka (version 1.12).
Two protein sequences of nitrofurantoin-resistant genes, nsfA and nsfB, were received from The Comprehensive Antibiotic Resistance Database (CARD, https://card.mcmaster.ca/) [15] and then aligned to No.8 genome and No.11 genome sequences via TBLASTN (0.05 e-value) in order to find matching sequences.
The assembled draft genomes of the five isolates were submitted to NCBI genome and SRA database (SAMN08287475, SAMN08287476, SAMN08287477, SAMN08287478 and SAMN0828747). The sequencing files were submitted to NCBI SRA database (SRP128497).
Six complete serotype III genomes were download from NCBI (NZ_CP022537.1/CP022537.1, NZ_CP010874.1/CP010874.1, NZ_CP010875.1/CP010875.1, NZ_CP011327.1/CP011327.1, NZ_CP011329.1/CP011329.1 and NC_004368.1/AL732656.1), respectively. The orthologous list of isolates No.8, No.11 and genomes downloaded from NCBI was built via orthoMCL (v1.4). The orthologous families which contain genes in isolates No.8, No.11 genome and in at most one complete genome downloaded from NCBI, genes in isolates No.8 genome and in at most one complete genome downloaded from NCBI, genes in isolates No.11 and in at most one complete genome downloaded from NCBI, genes in isolates No.8 and No.11 that did not be recorded to orthologous list were extracted by our own PERL script. We also built another orthologous list between isolates No.8 and No.11.
The orthologous list of isolates No.8, No.11, No.17, No.20 and No.24 was built via orthoMCL (v1.4). The orthologous families which contain genes both in isolates No.8 and No.11 genome were also extracted by our own PERL script.
Results
Group B Streptococcus can cause urinary tract infections (UTI). From November 2015 to March 2017 in Suzhou Hospital of Traditional Chinese Medicine, there were 147 individuals had positive urine culture of gram-positive bacteria and 30 (20.4%) of them were positive for GBS. However, few studies reported the GBS in urinary tract. Hence, we presented 27 GBS samples isolated from urine to investigate potential clinical treatment of GBS infections in Suzhou, China.
Serotypes
Five serotypes were found in 27 samples, which were Ia, Ib, III, V and VI. The proportion and number were 18.5% (5/27), 22.2% (6/27), 29.6% (8/27), 22.2% (6/27) and 7.4% (2/27) respectively (Table 1).
Resistance of tetracycline, erythromycin and clindamycin
All the 27 samples were susceptible to ceftriaxone, penicillin, vancomycin, linezolid, quinupristin–dalfopristin and tigecycline, while the highest percentage of 74.1% (20/27, Table 1 and Additional file 1: Table S6) samples were resistant to tetracycline. In previous studies, only the PCR primers of genes involved in resistance to tetracycline, erythromycin and clindamycin has been reported. In our PCR test of antibiotic resistance genes, tetM was the dominating resistance gene to tetracycline with 90.0% (18/20) occurrence. Other genes resistant to tetracycline were tetO, tetK and tetL, and the rates were 15.0% (3/20), 5.0% (1/20) and 5.0% (1/20) respectively.
The resistant rate measured for erythromycin was 63.0% (17/27). ErmB, mefE and ermTR were found and the rates are 47.1% (8/17), 41.2% (7/17) and 17.6% (3/17) respectively. About 44.4% (12/27) isolates were resistant to clindamycin. ErmB 75.0% (9/12), lnuB 58.3% (7/12) and ermTR 8.3% (1/12) were found in these isolates. Seven isolates were resistant to erythromycin but susceptible to clindamycin. Three of them were ‘inducible resistance to macrolides, lincosamides and streptograminB, (iMLSB) phenotype since they were positive in D test (Additional file 2: Figure S1). Four isolates were ‘resistance to macrolides and susceptibility to lincosamides (MS)’ phenotype. The erm (erythromycin ribosome methylase) gene has been considered as the reason of resistance to erythromycin, clindamycin and streptograminB, or the cMLSB. There were ten isolates resistant to erythromycin and clindamycin (cMLSB) with a high rate of ermB (80.0%) and lnuB (70.0%) detection. Five cMLSB isolates have both ermB and lnuB genes. Three cMLSB isolates have ermB gene without lnuB gene, two isolates have lnuB gene without ermB gene.
The PCR tests of common drug resistance genes (resistance genes for tetracycline, erythromycin and clindamycin) could explain 74.1% phenotype of drug resistances, except for the seven isolates (No.2, No.5, No.9, No.11, No.14, No.19 and No.25, Table 1 and Additional file 1: Table S6). We sequenced the genome of No.11 isolate and found another known erythromycin resistance gene, ermT (Table 3), suggesting that potentially there were other known resistance genes in the remaining six isolates.
Multiple resistance and detection for novel antibiotic resistance genes
Isolates of No.8, No.11, No.17 and No.24 were nonsusceptible to at least three antibiotics. As the control, No.20 isolate was susceptible to all antibiotics. These five isolates were sequenced. The draft genome sizes were from 2.06 to 2.20 MB, and the scaffold N50 s were from 107,763 to 468,051 bp (Table 2). As expected, none known resistance genes were found in PCR test and draft genome of No.20 isolate.
The No.8 isolate was resistant to tetracycline, erythromycin, clindamycin, and intermediate to nitrofurantoin. Genes tetO (tetracycline resistant), ermB (erythromycin and clindamycin resistant), mefE (erythromycin resistant) and lnuB (clindamycin resistant) were found in the PCR test. Genes tetO (tetracycline resistant), mefA (erythromycin resistant) and lnuB (clindamycin resistant) were found in the draft genome. No nitrofurantoin resistance gene was found (Tables 1, 3). No.11 isolate was resistant to tetracycline, erythromycin, and intermediate to nitrofurantoin. Tetracycline resistance gene tetM was found in PCR test and in the draft genome, but no erythromycin resistance gene was found in PCR test. An erythromycin and clindamycin resistance gene ermB was found in the draft genome. However, no nitrofurantoin resistance gene was found. About 48.1% (13/27, Additional file 1: Table S6) of the isolates were resistant to fluoroquinolones. One of them, No.17 isolate, was resistant to tetracycline, erythromycin and fluoroquinolones. Fluoroquinolones resistance gene was not included in the targets of PCR test, but the pmrA was found in the draft genome. The tetM (tetracycline resistant) was found in PCR test and in the draft genome. The ermTR (erythromycin and clindamycin resistant) and mefE (erythromycin resistant) were found in PCR test. The mefA (erythromycin resistant) was found in the draft genome. No.24 isolate was resistant to tetracycline, erythromycin and clindamycin. Tetracycline resistance genes tetM, tetK and tetL, erythromycin and clindamycin resistance gene ermB and clindamycin resistance gene lnuB were found in PCR test. Tetracycline resistance genes tetM and tetL and erythromycin resistance gene macB were found in the draft genome.
About 18.5% (5/27, Additional file 1: Table S6) of the isolates were intermediate to nitrofurantoin, among which four nitrofurantoin-intermediate isolates were serotype III and the other one was serotype Ib. So far, two nitrofurantoin-resistant genes, nsfA and nsfB, have been reported [16]. About two-thirds of the length of nsfB protein were aligned to all of the five draft genomes by TBLASTN search (E-value threshold 0.05, Additional file 1: Table S7), in which the genome area contained a ‘Ribosomal large subunit pseudouridine synthase B’ f in No.8 isolate, a ‘glyoxal reductase’ in No.11, a “putative NAD(P)H nitroreductase YfkO” in No.17, No.20 and No.24 isolates. So that the phenotype of nitrofurantoin intermediation was not caused by nsfB in our cases.
Based on the assumption that the mechanism of the nitrofurantoin intermediation in two isolates was similar, six complete serotype III genomes download from NCBI were included and built orthologous list with genomes of the nitrofurantoin-intermediate samples to search for candidate genes, given that the nitrofurantoin intermediation could not be explained by reported nitrofurantoin-resistant genes. Considering that a detailed information for nitrofurantoin resistance of these complete serotype III genomes is limited, we supposed that at most one of these genomes has a nitrofurantoin-resistant phenotype according to the reference resistance occurrence. Seventeen orthologous contain genes in genomes of No.8, No.11 isolates only or mixed with at most one complete genome downloaded from NCBI. There were 18 genes of isolate No.8 and 17 genes of isolate No.11 in these seventeen orthologous (Additional file 1: Table S8). Most of those genes (16 of 35) were annotated as “hypothetical proteins” since their biological function has still not been clarified. We also identified several genes associated with transposition (6 of 35) and bacteriolysis (2 of 35), which appear to be unessential for the nitrofurantoin intermediation. Therefore, the nitrofurantoin-resistant related genes probably exist in those “hypothetical proteins”. There were 27 genes distributed in the orthologous which contain genes only in isolate No.8 or mixed with at most one NCBI genome. 60 genes were distributed in the orthologous which contain genes only in isolate No.11 or mixed with at most one NCBI genome. Totally 148 genes of No.8 and 53 genes of No.11 were not recorded in this orthologous list. These genes cannot be found in another orthologous list which built between No.8 genome and No.11 genome either, suggesting that these 148 genes in No.8 and 53 genes in No.11 are unique genes of their own.
To further investigate the nitrofurantoin intermediation related genes in isolates No.8 and No.11 genome, the orthologous list of isolates No.8, No.11, No.17, No.20 and No.24 was built, with the other three genomes as the nitrofurantoin-susceptible control. A high proportion of genes were annotated as “hypothetical proteins” (10 of 28, Additional file 1: Table S9), which was consistent with the above-mentioned observation. On the basis of our findings, it can be predicted that there is a higher possibility for the intersection of genes identified in both two orthologous lists to be the candidate genes, which were classified as hypothetical protein, helix-turn-helix domain protein and recombinase. With the range of candidates narrowed down, the four genes annotated as hypothetical proteins (Additional file 1: Table S9) were more likely to be the nitrofurantoin-resistant related genes, providing valuable reference for future experimental study.
Discussion
Five serotypes were detected in our research, encompassing Ia, Ib, III, V and VI. The serotype III, V and Ib were of the highest proportion. The serotype III GBS was the most common type detected in our isolates, which was in line with studies from other countries [17, 18].
All of the GBS isolates studied in this work were susceptible to penicillin, indicating that penicillin is still a considerable choice for treatment of GBS infection. There was high rate of resistance to tetracycline (74.1%) in the test. However, this rate was lower than that reported in Beijing city (83.9%, isolated from vagina or rectum) [19], Taiwan (97.2%, isolated from urine, genital tract, genital tract, wound or pus, blood and respiratory tract) [20] and North American (89.2%, isolated from vagina or rectum) [17]. Compared to Shanghai, a city close to Suzhou, the rate of resistance to erythromycin in Suzhou (63.0%) is higher than reported in Shanghai (51.9%), however the rate of resistance to clindamycin in Suzhou (44.4%) is lower than that of Shanghai (61.5%) [21]. Note that, 80% of the nitrofurantoin-nonsusceptible GBSs in Suzhou were serotype III isolates, much higher than the 29.6% proportion of serotype III occurrence in the studied samples.
The tetM gene was the prevalent resistance gene (18/20) in tetracycline resistance isolates. The other detected tetracycline-nonsusceptible related genes were tetO, tetK and tetL. Besides, ermB (8/17) was the prevalent gene related to erythromycin resistance, which had been reported in Korea (isolated from vagina) [22]. It has been reported that the predominant genes in cMLSB were ermB in Spain [23, 24] and ermB and ermB-mef(A/F) in Shanghai city [21]. Interestingly, except ermB, we found lnuB also showed a high proportion (7/10) in cMLSB in Suzhou (70.0%), five isolates have both ermB and lnuB genes, three isolates have ermB gene only and two isolates have lnuB gene only. Although this high rate of lnuB had not been reported in previous studies.
We suspected that new resistance genes probably exist in the genomes of isolates No.8 and No.11 since the phenotype of nitrofurantoin intermediation in our cases was not caused by nsfA and nsfB, nitrofurantoin-resistant genes that were recorded in CARD database. If the nitrofurantoin resistance mechanism is similar in the two samples, the 35 genes in the orthologous which contain genes both in the genomes of isolates No.8 and No.11 only or mixed with at most one NCBI genome would be potential candidates for the nitrofurantoin resistance. We also built another orthologous list with the other three genomes of the five isolates as the nitrofurantoin-susceptible control to integrate our finding. There is a greater likelihood for the four genes annotated as hypothetical proteins in the intersection of the two orthologous lists to be the nitrofurantoin-resistant related genes.
Conclusions
We isolated 27 GBS samples from urine in Suzhou, China. Five serotypes were identified (Ia, Ib, III, V and VI). The serotype III was the primary type. All of the samples were susceptible to ceftriaxone, penicillin, vancomycin, linezolid, quinupristin–dalfopristin and tigecycline. The resistance rates to tetracycline, erythromycin, clindamycin and fluoroquinolones were 74.1, 63.0, 44.4 and 48.1% respectively. The resistance to tetracycline was mainly associated with the gene tetM (18/20). The erythromycin resistance was mainly associated with the genes ermB (8/17) and mefE (7/17). The genes ermB (8/10) and lnuB (7/10) occupied high rates in cMLSB phenotype. We found five samples were nonsusceptible to nitrofurantoin, and four of them were serotype III, among which two genomes of isolates do not contain any reported nitrofurantoin resistance genes. Five isolates genomes were sequenced and submitted to NCBI genomes database (SAMN08287475, SAMN08287476, SAMN08287477, SAMN08287478 and SAMN0828747) and SRA database (SRP128497).
Abbreviations
- GBS:
-
group B Streptococcus
- UTI:
-
urinary tract infections
- cMLSB:
-
constitutive resistance to macrolides, lincosamides, and streptograminB
- iMLSB:
-
inducible resistance to macrolides, lincosamides, and streptograminB
- MS:
-
resistance to macrolides and susceptibility to lincosamides
- CLSI:
-
clinical and laboratory standards institute guidelines
- NCBI:
-
National Center for Biotechnology Information
- ARDB:
-
Antibiotic Resistance Genes Database
- CARD:
-
Comprehensive Antibiotic Resistance Database
- PCR:
-
the polymerase chain reaction
- MIC:
-
minimal inhibit concentration
References
Delannoy CM, Crumlish M, Fontaine MC, Pollock J, Foster G, Dagleish MP, Turnbull JF, Zadoks RN. Human Streptococcus agalactiae strains in aquatic mammals and fish. BMC Microbiol. 2013;13:41. https://doi.org/10.1186/1471-2180-13-41.
Liu G, Zhang W, Lu C. Comparative genomics analysis of Streptococcus agalactiae reveals that isolates from cultured tilapia in China are closely related to the human strain A909. BMC Genomics. 2013;14:775. https://doi.org/10.1186/1471-2164-14-775.
Zuba S, de Villiers EP, Fuxelius HH, Andersson G, Johansson KE, Bishop RP, Bongcam-Rudloff E. Genome sequence of Streptococcus agalactiae strain 09mas018883, isolated from a Swedish cow. Genome Announc. 2013;1(4):e00456-13. https://doi.org/10.1128/genomeA.00456-13.
Poyart C, Tazi A, Réglier-Poupet H, Billoët A, Tavares N, Raymond J, Trieu-Cuot P. Multiplex PCR assay for rapid and accurate capsular typing of group B Streptococci. J Clin Microbiol. 2007;45(6):1985–8. https://doi.org/10.1128/JCM.00159-07.
Stoner TD, Weston TA, Trejo J, Doran KS. Group B streptococcal infection and activation of human astrocytes. PLoS ONE. 2015;10(6):e0128431. https://doi.org/10.1371/journal.pone.0128431.
Mhalu FS. Infection with Streptococcus agalactiae in a London hospital. J Clin Pathol. 1976;29(4):309–12. https://doi.org/10.1136/jcp.29.4.309.
Porta K, Rizzolo D. Preventing group B streptococcal infections in newborns. J Am Acad Phys. 2015;28(3):24–9. https://doi.org/10.1097/01.JAA.0000460915.52391.70.
Ulett KB, Benjamin WH, Zhuo FL, Xiao M, Kong FR, Gilbert GL, Schembri MA, Ulett GC. Diversity of group B Streptococcus serotypes causing urinary tract infection in adults. J Clin Microbiol. 2009;51(8):2806. https://doi.org/10.1128/JCM.01067-13.
Trivalle C, Martin E, Martel P, Jacque B, Menard JF, Lemeland JF. Group B streptococcal bacteraemia in the elderly. J Med Microbiol. 1998;47(7):649–52. https://doi.org/10.1099/00222615-47-7-649.
Church D, Carson J, Gregson D. Point prevalence study of antibiotic susceptibility of genital group B Streptococcus isolated from near-term pregnant women in Calgary, Alberta. Can J Infect Dis Med Microbiol. 2012;23(3):121–4. https://doi.org/10.1155/2012/876103.
Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing. Clinical and Laboratory Standards Institute: Wayne; 2017 (M100-S27).
Seppälä H, Nissinen A, Yu Q, Huovinen P. Three different phenotypes of erythromycin-resistant Streptococcus pyogenes in Finland. J Antimicrob Chemother. 1993;32(6):885–91. https://doi.org/10.1093/jac/32.6.885.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77. https://doi.org/10.1089/cmb.2012.0021.
Liu B, Pop M. ARDB—antibiotic resistance genes database. Nucleic Acids Res. 2009;37:D443–7. https://doi.org/10.1093/nar/gkn656.
Jia BF, Raphenya AR, Alcock B, Waglechner N, Guo PY, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–73. https://doi.org/10.1093/nar/gkw1004.
Shanmugam D, Esak SB, Narayanaswamy A. Molecular characterisation of nfsA gene in nitrofurantoin resistant uropathogens. J Clin Diagn Res. 2016;10(6):DC05–9. https://doi.org/10.7860/JCDR/2016/17280.7957.
Teatero S, Ferrieri P, Martin I, Demczuk W, McGeer A, Fittipaldi N. Serotype distribution, population structure, and antimicrobial resistance of group B Streptococcus strains recovered from colonized pregnant women. J Clin Microbiol. 2017;55(2):412–22. https://doi.org/10.1128/JCM.01615-16.
Mengist A, Kannan H, Abdissa A. Prevalence and antimicrobial susceptibility pattern of anorectal and vaginal group B Streptococci isolates among pregnant women in Jimma, Ethiopia. BMC Res Notes. 2016;91:351. https://doi.org/10.1186/s13104-016-2158-4.
Wang P, Tong JJ, Ma XH, Song FL, Fan L, Guo CM, Shi W, Yu SJ, Yao KH, Yang YH. Serotypes, antibiotic susceptibilities, and multi-locus sequence type profiles of Streptococcus agalactiae isolates circulating in Beijing, China. PloS ONE. 2015;10(3):e0120035. https://doi.org/10.1371/journal.pone.0120035.
Ko WC, Lee HC, Wang LR, Lee CT, Liu AJ, Wu JJ. Serotyping and antimicrobial susceptibility of group B Streptococcus over an eight-year period in southern Taiwan. Eur J Clin Microbiol Infect Dis. 2001;20(5):334–9. https://doi.org/10.1007/s10096-001-8115-7.
Yan YZ, Hu H, Lu TY, Fan HQ, Hu Y, Li G, Zhang XH, Shi Y, Xia R. Investigation of serotype distribution and resistance genes profile in group B Streptococcus isolated from pregnant women: a Chinese multicenter cohort study. APMIS. 2016;124(9):794–9. https://doi.org/10.1111/apm.12570.
Seo YS, Srinivasan U, Oh KY, Shin JH, Chae JD, Kim MY, Yang JH, Yoon HR, Miller B, DeBusscher J. Changing molecular epidemiology of group B Streptococcus in Korea. J Korean Med Sci. 2010;25(6):817–23. https://doi.org/10.3346/jkms.2010.25.6.817.
Gonzalez JJ, Andreu A. Multicenter study of the mechanisms of resistance and clonal relationships of Streptococcus agalactiae isolates resistant to macrolides, lincosamides, and ketolides in Spain. Antimicrob Agents Chemother. 2005;49(6):2525–7.
Betriu C, Culebras E, Gómez M, Rodríguez-Avial I, Sánchez BA, Agreda MC, Picazo JJ. Erythromycin and clindamycin resistance and telithromycin susceptibility in Streptococcus agalactiae. Antimicrob Agents Chemother. 2003;47(3):1112–4. https://doi.org/10.1128/AAC.47.3.1112-1114.2003.
Authors’ contributions
GY collected the GBS samples, carried out the serotyping and drug testing experiment and sequenced genomes. DX carried out the bioinformatics analysis and drafted the manuscript. LY and ZL helped to draft the manuscript. ZGP raised useful suggestions. ZY conceived of the study, participated in its design and coordination, helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank the Suzhou Hospital of Traditional Chinese Medicine kindly provided the GBS samples for this study.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Six complete serotype III genomes were download from NCBI (NZ_CP022537.1/CP022537.1, NZ_CP010874.1/CP010874.1, NZ_CP010875.1/CP010875.1, NZ_CP011327.1/CP011327.1, NZ_CP011329.1/CP011329.1 and NC_004368.1/AL732656.1, https://www.ncbi.nlm.nih.gov/genome). The genome sequencing data is submitted to NCBI genomes database (SAMN08287475, SAMN08287476, SAMN08287477, SAMN08287478 and SAMN0828747, https://www.ncbi.nlm.nih.gov/genome) and SRA database (SRP128497, https://www.ncbi.nlm.nih.gov/sra). Other data that generated or analysed during this study are included in this published article.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work was funded in part by the National Key Research Program of China (2017YFC0907503), Shanghai Municipal Science and Technology Commission (16ZR1423900 and 17DZ2270800).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
The primers of serotype testing PCR. Table S2. The PCR amplification system of cps gene. Table S3. The procedure of PCR amplification system for cps gene. Table S4. The PCR amplification products of serotype Ia to IX. Table S5. The primers of drug resistance genes. Table S6. Antimicrobial susceptibility test result. Table S7. TBLAST result of nsfB against five draft genomes. Table S8. The function and numbers of genes in orthologous which contain genes in genomes of isolates both No.8 and No.11 only or mixed with at most one NCBI genome. Table S9. The function and numbers of genes in orthologous which contain genes in genomes of isolates No.8, No.11, No.17, No.20 and No.24.
Additional file 2: Figure S1.
Positive results (iMLSB) in D-test. From left to right are No.10, No.11 and No.17, respectively.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Guo, Y., Deng, X., Liang, Y. et al. The draft genomes and investigation of serotype distribution, antimicrobial resistance of group B Streptococcus strains isolated from urine in Suzhou, China. Ann Clin Microbiol Antimicrob 17, 28 (2018). https://doi.org/10.1186/s12941-018-0280-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12941-018-0280-y