
Abstract
Background
Plasmodium vivax is the most widespread human malaria parasite outside Africa and is the predominant parasite in the Americas. Increasing reports of P. vivax disease severity, together with the emergence of drug-resistant strains, underscore the urgency of the development of vaccines against P. vivax. Polymorphisms on DBP-II-gene could act as an immune evasion mechanism and, consequently, limited the vaccine efficacy. This study aimed to investigate the pvdbp-II genetic diversity in two Brazilian regions with different epidemiological patterns: the unstable transmission area in the Atlantic Forest (AF) of Rio de Janeiro and; the fixed malaria-endemic area in Brazilian Amazon (BA).
Methods
216 Brazilian P. vivax infected blood samples, diagnosed by microscopic examination and PCR, were investigated. The region flanking pvdbp-II was amplified by PCR and sequenced. Genetic polymorphisms of pvdbp-II were estimated based on the number of segregating sites and nucleotide and haplotype diversities; the degree of differentiation between-regions was evaluated applying Wright’s statistics. Natural selection was calculated using the rate of nonsynonymous per synonymous substitutions with the Z-test, and the evolutionary distance was estimated based on the reconstructed tree.
Results
79 samples from AF and 137 from BA were successfully sequenced. The analyses showed 28 polymorphic sites distributed in 21 codons, with only 5% of the samples Salvador 1 type. The highest rates of polymorphic sites were found in B- and T cell epitopes. Unexpectedly, the nucleotide diversity in pvdbp-II was higher in AF (0.01) than in BA (0.008). Among the 28 SNPs detected, 18 are shared between P. vivax isolates from AF and BA regions, but 8 SNPs were exclusively detected in AF—I322S, K371N, E385Q, E385T, K386T, K411N, I419L and I419R—and 2 (N375D and I419M) arose exclusively in BA. These findings could suggest the potential of these geographical clusters as population-specific-signatures that may be useful to track the origin of infections. The sample size should be increased in order to confirm this possibility.
Conclusions
The results highlight that the pvdbp-II polymorphisms are positively selected by host’s immune pressure. The characterization of pvdbp-II polymorphisms might be useful for designing effective DBP-II-based vaccines.
Similar content being viewed by others


Background
Plasmodium vivax is the most widespread human malaria parasite outside sub-Saharan Africa. Globally, 7.5 million cases in 2017 were caused by P. vivax, that is responsible for 37% of malaria cases in South-East Asia, 31% in the Eastern Mediterranean, and 74% in the Americas [1]. Plasmodium vivax causes significant morbidity, as well as social and economic burden, becoming a public health challenge in endemic countries [2]. The evidence of severe vivax malaria around the world, including Brazil [3], together with the emergence of drug-resistant strains [4], underscore the urgency to reduce the infection burden and malaria elimination [5, 6]. Therefore, the development of vaccines that protect against P. vivax blood stages is a priority to prevent disease and onward transmission.
The Duffy Binding Protein–DBP, a 140-kDa protein expressed in the micronemes of the P. vivax surface in the asexual blood-stage, interacts with the Duffy antigen/receptor for chemokines (DARC) on the host erythrocytes [7], making this molecule an attractive vaccine candidate against vivax malaria.
Currently, there are ongoing studies on pvdbp-II diversity. In Brazil, the unique study in this sense with Brazilian Amazon Basin samples showed codons evolving under natural selection [8]. The present study now investigates, besides Brazilian Amazon, the Rio de Janeiro Atlantic Forest region (AF) that present peculiar epidemiological characteristics [9].
Methods
Sample collection, diagnosis, and DNA extraction
Brazil, a country of continental proportions, has three malaria transmission profiles. The first and most important occurs inside the Brazilian Amazon Basin (BA), where more than 99% of malaria cases were recorded; the second one involves imported malaria cases, which corresponds to infections that are acquired in Brazilian endemic areas different from where the individual lives or the diagnosis has been done, or from other endemic countries. The third type of transmission represents around 0.05% of all malaria cases in Brazil and corresponds to autochthonous malaria in the Atlantic Forest (AF), located primarily along the south-eastern Atlantic Coast [9].
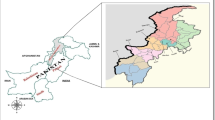
The polymorphisms in pvdbp-II were investigated in samples representing these three malaria profiles observed in Brazil. 227 P. vivax samples diagnosed by thin and thick blood smear and PCR were collected [13]. Two groups of patients were analysed. One group of 177 individuals (95 from BA and 82 from AF) who attended to the Reference Centre for Malaria Diagnosis CPD-Mal/Fiocruz in Rio de Janeiro (S 22° 54′ W 43° 12′) from January 2011 to March 2018; the other group of 50 individuals who sought for diagnosis at the Unit Health of Tucuruí (S 3º 46′ W 49º 40′), a municipality from Pará State, located in the Brazilian Amazon region during 2011. The samples were separated according to the state of infection (Fig. 1) and the year of blood collection (Table 1). The study was approved by the Ethics Research Committee of Instituto Oswaldo Cruz, Fiocruz, Brazil (69256317.3.0000.5248). All volunteers’ patients signed a written informed consent before collection of 4 mL of venous blood. The DNA from 1 mL blood samples was extracted using QIAamp™ DNA Blood Midi Kit (QIAGEN), according to the manufacturer’s instructions.

Brazilian endemic areas according to the risk (Annual Parasitic Incidence–API) (SIVEP-Malaria). Sampling regions of Rio de Janeiro Atlantic Forest and states of the Brazilian Amazon region circumscribed in blue
pvdbp-II amplification
The PCR reaction to amplify a fragment of 675 bp of the DBP-II-encoding gene (amino acids from 290 to 515) was carried out with the primers 5′ ATGTATGAAGGAACTTACGAAT 3′ (forward) and 5′ ACCTGCCGTCTGAACCTTTT 3′ (reverse), as previously described [8]. The 20 μL of reaction mixture contained 2 μL (100–200 ng) of DNA, 1 μM primers (forward and reverse) and 5 × HOT FIREPol™ Blend Master Mix (Solis Biodyne), as a source of DNA polymerase, 1 mM of dNTP, 12.5 mM of MgCl2 and enzyme buffer (saline solution), including dyes to increase sample density during the agarose gel electrophoresis. The amplification cycles comprised: 95 ℃ for 12 min, 30 cycles of 95 ℃ for 30 s, 54 ℃ for 45 s and 72 ℃ for 2 min, followed by an extension cycle of 72 ℃ for 10 min. PCR products were purified using the kit Wizard™ SV Gel and PCR Clean-Up System (Promega), following the manufacturer’s procedure. Then, the purified forward and reverse strands were subjected to cycle sequencing with the Big Dye™ Terminator Cycle Sequencing Ready Reaction version 3.1 (Applied Biosystems) using the PCR primers at a concentration of 3.2 μM. The DNA sequencing was performed in ABI Prism DNA Analyzer™ 3730 (Applied Biosystems) with the support of Fiocruz Genomic Platform RPT01A. The sequenced reads were first analysed using NovoSNP software to investigate polymorphisms; the cutoff of electropherogram quality score was 10 to avoid losing some variations. Also, sequences were analysed in BioEdit sequence alignment editor to better-visualized SNP positions, employing ClustalW multiple sequence aligner. The Salvador 1 (Sal-1) strain was used as a reference sequence (PVX_110810, from PlasmoDB: http://www.plasmoDB.org). Sequences displaying singleton mutations and/or overlapped peaks after chromatogram inspection were re-sequenced. A single infection or the predominant variant was considered when only one nucleotide peak (allele), at any polymorphic locus of the sequence, was observed.
Genetic diversity, natural selection, and statistical analysis
Genetic diversity of pvdbp-II sequences was analysed using the DnaSP 6.11 software [10] to estimate within-population diversity based on the number of segregating sites (S) and nucleotide (π) and haplotype (Hd) diversities. Wright’s fixation statistics (F) determined the degree of differentiation between-populations [11]. Evolutionary analyses were conducted with MEGA7 v7.0 software [12]. The neutrality test of evolution was calculated by Z-test using the Nei-Gojobori method [13], in which the rate of the average number of non-synonymous (dN) and synonymous (dS) SNPs define if the selective pressure is positive (dN > dS), negative (dN < dS) or neutral (dN = dS). Variance differences, in both AF and BA sequence datasets, were computed using the bootstrap method (1000 replicates). Evolutionary distances were calculated using the p-distance method [14], and the pvdbp-II SNP-based tree was reconstructed using the Neighbour-joining method [15] and bootstrap analysis (500 replicates) to measure accuracy. Graphs were building using GraphPad Prism software 8.1.2.
Results
Among the 227 samples collected, 216 (95%) had the 675 bp pvdbp-II fragment amplified: 79 from Rio de Janeiro AF and 137 from North BA region, comprising the states of Pará (50), Amazonas (48), Acre (11), Roraima (3), Rondônia (17), and Amapá (8). The failure to satisfactory amplify 11 samples might be somehow attributed to primer limitations due to unknown polymorphisms in target sequences.
As no remarkable differences were found among P. vivax parasite populations from different BA states, pvdbp-II polymorphism will be presented, regardless of the BA state where the samples were collected. Concerning temporal differences of pvdbp-II polymorphism in isolates collected in the same area at different time, only in Amazonas state–São Gabriel da Cachoeira locality—several representative samples could be obtained on different occasions. In this case, temporal differences seem to occur randomly, independently of the year, and the number of haplotypes was proportional to the number of samples; the same was true in AF samples, but here the turn-over of parasite populations including identical haplotypes along the years was higher. However, as the sampling number of AF is much higher than in São Gabriel da Cachoeira, no definitive conclusions should be made (Additional file 1).
In all 216 Brazilian samples, the comparison of the pvdbp-II amplification products with the Sal-1 type strain revealed 28 polymorphic sites of which one was synonymous and 27 non-synonymous. These 28 SNPs spanned 21 codons (Table 2). Of these polymorphic codons, 19 presented only one base substitution and two codons—385 (G1153A and A1154C), and 386 (A1157C and G1158T)–presented substitutions in two bases. The 79 AF samples had 26 SNPs, of which 25 were non-synonymous. The 137 BA samples contained 20 SNPs, of which 19 are non-synonymous (Additional file 2). The AF and BA regions shared 18 SNPs, and the majority of these polymorphisms showed higher frequencies in AF (11 codons) than in BA (six codons) regions, while one codon has the same frequency in both regions (Fig. 2 and Table 2).

Pvdbp-II codons in Brazilian isolates from two Brazilian geographic regions: AF (79) and BA (137). The SNP identification code represents the amino acid in the Sal-1 reference sequence (first character), followed by the position of this residue (number), and the replacing amino acid observed at the same position (last character); Atlantic Forest from Rio de Janeiro (AF); Brazilian Amazon (BA); N Number, SNPs Single Nucleotide Polymorphisms. The graph was constructed using GraphPad Prism software 8.1.2
Overall, D384G (191/88%), L424I (152/70%), and R390H (113/52%) SNPs were the most frequent. In AF region, D384G was identified in all but one sample (99%; 78), L424I in 97% (77), and R390H in 75% (59) of the samples. In BA region, D384G was present in 82% (113), L424I in 55% (75), and R390H in 39% (54) of the samples. SNPs I322S (17/22%), K371N (10/13%), E385Q (21/27%), E385T (3/4%), K386T (22/28%), K411N (60/76%), I419L (20/25%), and I419R (6/8%) were exclusively detected in AF while N375D (6/4%) and I419M (13/9%) were exclusively found in BA (Fig. 2).
The nucleotide diversity of pvdbp-II from AF (π = 0.011) was higher than those from BA (π = 0.008) (Table 3). The non-synonymous substitutions exceeded the synonymous ones (Dn > Ds) with significant expressive values in all Brazilian isolates (2.40; < 0.009), as well as when segregated AF (2.02; < 0.02) and BA (3.10; < 0.001) regions (Table 3).
Polymorphic sites and nucleotide diversity were also investigated in eight peptides previously identified in pvdbp-II B- and T-cells epitopes [16, 17]. The highest number of polymorphic sites in overall Brazilian samples were found in B cell epitopes: four nsSNPs in peptides 45 (384, 385, 386, and 390) and 48 (385, 386, 390, and 398), revealing a nucleotide diversity of π = 0.051 and π = 0.047, respectively, followed by T-cell epitope in peptide 66 (π = 0.020) with one nsSNP and peptide 5 (π = 0.011) with two nsSNPs. Peptides 16 (π = 0.009), 20 (π = 0.008), 78 (π = 0.005) and 13 (π = 0.003), had only one nsSNP each one of them (Table 4). The peptides 45, 48 and 66 concentrated the higher nucleotide diversity of the pvdbp-II fragment.
The simultaneous presence of four SNPs in peptide 45 coding region was detected in 72% of AF isolates and only 9% of BA, whereas in peptide 48, only 1% of AF and 4% of BA showed simultaneous presence of three SNPs.
Considering that N417K, L424I, W437R, or I503K mutations could affect pvdbp-II antigenicity, the presence of such polymorphisms had been sought. The N417K—L424I—W437R combination was found in 35% (81) of the samples: 20% (16) in AF and 42% (58) in BA. Among this combination, 19% (44) of the samples bear the fourth polymorphic allele I503K: 9% (seven) in AF and 23% (32) in BA.
pvdbp-II haplotypes
Among the 216 Brazilian isolates, only 11 (5%) have sequences identical to Sal-1 type (NRILKNRDEKRSTKNILWQQI): one sample from AF and ten from BA. The allele’s pattern comprised 90 haplotypes, and 59 of them (66%) were detected in one single parasite isolate each. The most polymorphic haplotypes had 15 SNPs and were only detected in AF isolates (4/6%).
DB01 (nine SNPs) and DB02 (three SNPs) were the more frequent haplotypes: DB01 was found exclusively in AF (16/20%) and DB02 exclusively in BA (16/12%) (Fig. 3), (Additional file 3). In AF, 36 haplotypes were found, and in BA 60. Of the haplotypes found in AF samples, 30 were exclusive of this area, and of the haplotypes found in BA, 54 were exclusive of this region.

Haplotypes (DB01-DB32) presented in more than one sample from Brazilian Amazon and Atlantic Forest. BA Brazilian Amazon samples with 89 (65%) samples presenting 22 haplotypes (DB02, DB03, DB04, DB05, DB06, DB08, DB09, DB10, DB12, DB13, DB14, DB15, DB16, DB19, DB20, DB21, DB22, DB28, DB29, DB30, DB31 and DB32). AF Atlantic Forest from Rio de Janeiro isolates with 58 (73%) samples showing 16 haplotypes (DB01, DB03, DB04, DB07, DB08, DB11, DB12, DB16, DB17, DB18, DB21, DB23, DB24, DB25, DB26 and DB27). The graph was constructed using GraphPad Prism software 8.1.2
The levels of haplotype diversity (Hd) were quite similar in AF (0.94) and BA (0.97).
The phylogenetic tree based on pvdbp-II SNPs revealed that parasites of the same geographic region shared similar profiles with few exceptions (Fig. 4). As expected, the AF samples due to its restrict geographic area were more clustering than BA. The AF samples were more distant to the Sal-1 reference strain than the BA samples. A significant degree of genetic differentiation (Fst = 0.36) was verified between AF and BA isolates.

Neighbour-Joining phylogenetic tree based on pvdbp-II-gene SNPs of Brazilian Amazon (137) and Atlantic Forest (79) samples
Discussion
This study was the first to investigate the genetic diversity of pvdbp-II outside the Amazon region, precisely in an unstable transmission area in the Atlantic Forest of Rio de Janeiro. Besides AF, pvdbp-II polymorphisms were also investigated in P. vivax isolates from the most significant Brazilian endemic region to expand the knowledge on parasite population diversity in Brazil and compare the data with those from other endemic countries. In this way, only 5% of field samples presented the pvdbp-II sequence utterly identical to the pvdbp-II of Salvador 1 type strain. The polymorphism degree comprised 28 polymorphic sites in 21 codons, with only one sSNP. The eighteen nsSNPs found in P. vivax isolates here studied, including those with higher frequencies—D384G and L424I were previously reported in P. vivax isolates from Africa, Oceania, Asia, and other South America countries [18,19,20,21]. The frequency of these SNPs was similar to this study, as in the case of Thailand [18], Myanmar [19], Sudan [21], and Papua New Guinea [17], or smaller as in the case of Sri-Lanka [22]. These data reinforce the idea that pvdbp-II diversity seems to occur independently of the malaria endemicity levels [23, 24].
Further, eight SNPs were exclusively detected in AF, and, interestingly, two of them (K371N and E385Q/K) had been previously found in AF areas of Santa Catarina state [25]. Likewise, two previously described SNPs (N375D and I419M) in the Brazilian Amazon region [26] and Asia [18, 19], arose exclusively in BA. These findings could suggest the potential of AF geographical clusters as population-specific-signatures that may be useful to track the origin of infections. However, more studies are required to confirm that these SNPs are confined to the determined geographic area.
Overall, the SNP-based phylogenetic tree of the pvdbp-II revealed two genetic subdivisions, one for AF and another for BA, showing that same geographic region isolates share similar evolutionary histories. BA samples are spread out in several clades, while AF samples show a small number of clades, possibly due to the tremendous difference in territorial size between these two geographic regions. The genetic subdivision of P. vivax Brazilian populations was also supported by a high Fst value [17], reflecting the limited gene flow between parasites of AF and BA. The high frequency of exclusive nsSNP K411N present in 76% of the isolates of AF could demonstrate the fixation of this SNP, probably, by positive natural selection on pvdbp-II mediated by host pressures. Inclusive, the hypothesis of positive selection could be sustained by the increased number of non-synonymous substitutions (Dn > Ds) assessed by the Z-test.
It is well known that nsSNPs could change B and/or T cell epitopes, therefore affecting the host immune response. In this study, the nucleotide diversity was higher in B and/or T cell epitopes than in the whole 675 bp amplified fragmented, highlighting a positive selection mediated by host immune system, that was supported by significant positive values in neutrality test. Except for I322S nsSNPs, exclusively detected in AF isolates, all other epitope polymorphisms were already reported in Brazil [8], Papua New Guinea [17], Thailand [20], Colombia and South Korea [17], demonstrating the global distribution of these epitope mutated alleles.
BA is a Brazilian region where malaria transmission effectively occurs, and much more meiotic recombination generating polymorphism is expected. Nevertheless, the SNP frequency in AF was higher than in BA, suggesting that besides recombination, the host immune response is an essential natural selection factor promoting fixation of mutated pvdbp-II parasite populations.
Conclusion
The results highlight that the pvdbp-II polymorphisms are positively selected by host’s immune response pressure. The characterization of pvdbp-II polymorphisms might be useful for designing effective DBP-II-based vaccines.
Availability of data and materials
All data generated or analysed during this study are included in this article (and its Additional files). Sequence data that support the findings of this study have been deposited in GenBank with the accession codes MN223747 to MN223974.
Abbreviations
- WHO:
-
World Health Organization
- Pvdbp-II:
-
Plasmodium vivax duffy binding protein-region II
- DARC:
-
Duffy antigen/receptor for chemokines
- AF:
-
Atlantic Forest
- BA:
-
Brazilian Amazon
- π:
-
Nucleotide diversity
- S:
-
Polymorphic sites
- Hd:
-
Haplotype diversity
- Fst:
-
Wright’s fixation statistics
- SNP:
-
Single nucleotide polymorphism
- sSNP:
-
Synonymous single nucleotide polymorphism
- nsSNP:
-
Non-synonymous single nucleotide polymorphism
References
WHO. World malaria report 2018. Geneva: World Health Organization; 2018. https://www.who.int/malaria/publications/world-malaria-report-2018/en/. Accessed 21 July 2019.
WHO. Confronting Plasmodium vivax malaria. Geneva: World Health Organization; 2015. http://www.who.int/malaria. Accessed 21 July 2019.
Lacerda MVG, Mourão MPG, Alexandre MA, Siqueira AM, Magalhães BML, Martinez-Espinosa FE, et al. Understanding the clinical spectrum of complicated Plasmodium vivax malaria: a systematic review on the contributions of the Brazilian literature. Malar J. 2012;11:12.
de Santana Filho FS, Lima Arcanjo AR, Chehuan YM, Costa MR, Martinez-Espinosa FE, Vieira JL, et al. Chloroquine-resistant Plasmodium vivax, Brazilian Amazon. Emerg Infect Dis. 2007;13:1125–6.
Auburn S, Böhme U, Steinbiss S, Trimarsanto H, Hostetler J, Sanders M, et al. A new Plasmodium vivax reference sequence with improved assembly of the subtelomeres reveals an abundance of pir genes. Wellcome Open Res. 2016;1:4.
Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM. Vivax malaria: neglected and not benign. Am J Trop Med Hyg. 2007;77:79–87.
Singh SK, Singh AP, Pandey S, Yazdani SS, Chittnis CE, Sharma A. Definition of structural elements in Plasmodium vivax and P. knowlesi Duffy-binding domains necessary for erythrocyte invasion. Biochem J. 2003;374:193–8.
Sousa TN, Tarazona-Santos EM, Wilson DJ, Madureira AP, Falcão PRK, Fontes CJF, et al. Genetic variability and natural selection at the ligand domain of the Duffy binding protein in Brazilian Plasmodium vivax populations. Malar J. 2010;9:334.
De Pina-costa A, Brasil P, Maria S, Santi D, De Araujo MP, Suárez-mutis MC, et al. Malaria in Brazil: what happens outside the Amazonian endemic region. Mem Inst Oswaldo Cruz. 2014;109:618–33.
Rozas J, Ferrer-Mata A, Carlos anchez-DelBarrio JS, Guirao-Rico S, Librado P, Ramos-Onsins SE, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34:3299–302.
Hudson RR. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 2002;18:337–8.
Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:18–26.
Nei M, Kumar S. Molecular evolution and phylogenetics. New York: Oxford University Press; 2000.
Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25.
Xainli J, Baisor M, Kastens W, Bockarie M, Adams JH, King CL. Age-dependent cellular immune responses to Plasmodium vivax Duffy binding protein in humans. J Immunol. 2002;169:3200–7.
Cole-Tobian J, King CL. Diversity and natural selection in Plasmodium vivax Duffy binding protein gene. Mol Biochem Parasitol. 2003;127:121–32.
Gosi P, Khusmith S, Khalambaheti T, Lanar DE, Schaecher KE, Fukuda MM, et al. Polymorphism patterns in Duffy-binding protein among Thai Plasmodium vivax isolates. Malar J. 2008;7:112.
Ju H-L, Kang J-M, Moon S-U, Kim J-Y, Lee H-W, Lin K, et al. Genetic polymorphism and natural selection of Duffy binding protein of Plasmodium vivax Myanmar isolates. Malar J. 2012;11:60.
Chootong P, McHenry AM, Ntumngia FB, Sattabongkot J, Adams JH. The association of Duffy binding protein region II polymorphisms and its antigenicity in Plasmodium vivax isolates from Thailand. Parasitol Int. 2014;63:858–64.
Hoque MR, Mohieldien M, Elfaki A, Ahmed A, Lee S-K, Muh F, et al. Diversity pattern of Duffy binding protein sequence among Duffy-negatives and Duffy-positives in Sudan. Malar J. 2018;17:297.
Premaratne PH, Aravinda BR, Escalante AA, Udagama PV. Genetic diversity of Plasmodium vivax Duffy binding protein II (Pvdbp-II) under unstable transmission and low intensity malaria in Sri Lanka. Infect Genet Evol. 2011;11:1327–39.
De Sousa TN, Carvalho LH, de Brito CFA. Worldwide genetic variability of the duffy binding protein: insights into Plasmodium vivax vaccine development. PLoS ONE. 2011;6:e22944.
George MT, Schloegel JL, Ntumngia FB, Barnes SJ, King CL, Casey JL, et al. Identification of an immunogenic broadly inhibitory surface epitope of the Plasmodium vivax Duffy binding protein ligand domain. mSphere. 2019;4:e00194.
Camargos Costa D, Pereira de Assis GM, de Souza Silva FA, Araújo FC, de Souza Junior JC, Braga Hirano ZM, et al. Plasmodium simium, a Plasmodium vivax-related malaria parasite: genetic variability of Duffy binding protein II and the Duffy antigen/receptor for chemokines. PLoS ONE. 2015;10:0131339.
De Sousa TN, Kano FS, De Brito CFA, Carvalho LH. The Duffy binding protein as a key target for a Plasmodium vivax vaccine: lessons from the Brazilian Amazon. Mem Inst Oswaldo Cruz. 2014;109:25–30.
Acknowledgements
We want to thank all the patients for agreeing to participate in this study and the health staff for helping with data collection and microscopic diagnosis. We also thank the staff from the Genomic Platform for DNA sequencing facilities RPT01A/PDTIS/Fiocruz. To CNPq, FAPERJ, CAPES, DECIT, Brazilian Malaria Control Program/SVS/MS, and Fiocruz by mainly funding the project.
Funding
NKAO is recipient of a Doctoral Fellowship from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES); RAF is recipient of a Scientific Initiation with support of Scholarship from the Fiocruz/Centro de Integração Empresa/Escola (CIEE); PB, CTDR and MFFC are recipients of Research Productivity Fellowships from the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq); PB, CTDR and MFFC are “Cientistas do Nosso Estado” from the Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado de Rio de Janeiro (FAPERJ).
Author information
Authors and Affiliations
Contributions
NKAO, RAF, LLC, and ARL performed experimental development and data analyses. PB conducted the clinical follow up with the patients. APC collected samples and epidemiological data. DP assisted with genetic diversity and statistical analyses. MC performed sequence and phylogenetic analyses. CTDR and MFFC revised the manuscript. NKAO and MFFC were involved in the design and planning of the study and preparation of the manuscript. CTDR, MFFC, and MC revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Research Committee of Oswaldo Cruz Institute, Fiocruz, Brazil. Reference number CAEE: 69256317.3.0000.5248.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Pvdbp-II-gene haplotypes in São Gabriel da Cachoeira (BA) and in Rio de Janeiro (AF).
Additional file 2.
Multiple sequence alignment of pvdbp-II from AF and BA regions. Multiple alignment of 675 bp pvdbp-II fragment of field isolates compared to Salvador 1 reference sequence (PVX_110810).
Additional file 3.
Pvdbp-II haplotypes. Identification of 90 haplotypes and their respective nucleotide sequence compared to Salvador 1 reference sequence (PVX_110810).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Almeida-de-Oliveira, N.K., Lima-Cury, L., de Abreu-Fernandes, R. et al. Extensive genetic diversity of Plasmodium vivax dbp-II in Rio de Janeiro Atlantic Forest and Brazilian Amazon Basin: evidence of positive selection. Malar J 19, 81 (2020). https://doi.org/10.1186/s12936-020-03159-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-020-03159-y