
Abstract
Background
Poor knowledge on the afebrile Plasmodium falciparum biology limits elimination approaches to target asymptomatic malaria. Therefore, the association of parasite factors involved in cytoadhesion, parasite multiplication and gametocyte maturation with afebrile malaria was assessed.
Methods
Plasmodium falciparum isolates were collected from febrile (axillary temperature ≥ 37.5 °C or a reported fever in the previous 24 h) and afebrile (fever neither at the visit nor in the previous 24 h) individuals residing in Southern Mozambique. var, PfSir2a and Pfs25 transcript levels were determined by reverse transcriptase quantitative PCRs (RT-qPCRs) and compared among 61 pairs of isolates matched by parasite density, age and year of sample collection.
Results
The level of varC and PfSir2a transcripts was higher in P. falciparum isolates from afebrile individuals (P ≤ 0.006), while varB and DC8 genes (P ≤ 0.002) were higher in isolates from individuals with febrile infections. After adjusting the analysis by area of residence, doubling the relative transcript unit (RTU) of varC and PfSir2a was associated with a 29.7 (95% CI 4.6–192.3) and 8.5 (95% CI 1.9–32.2) fold increases, respectively, of the odds of being afebrile. In contrast, doubling the RTU of varB and DC8 was associated with a 0.8 (95% CI 0.05–0.6) and 0.2 (95% CI 0.04–0.6) fold changes, respectively, of the odds of being afebrile. No significant differences were found for Pfs25 transcript levels in P. falciparum isolates from afebrile and febrile individuals.
Conclusions
var and gametocyte-specific transcript patterns in febrile and afebrile infections from southern Mozambique matched by age, parasite density and recruitment period suggest similar transmissibility but differential expression of variant antigens involved in cytoadhesion and immune-evasion.
Similar content being viewed by others


Background
Asymptomatic Plasmodium falciparum infections debilitate the health of affected population while representing a hidden source of continuous transmission that can compromise elimination efforts [1]. Poor knowledge on the dynamics, transmissibility and biological mechanisms of afebrile infections restrict the evidence-based approaches to deal with this hidden reservoir.
Transmissibility and clinical impact of P. falciparum infections is dependent on gametocyte production and parasite virulence [1]. Higher carriage of gametocyte stages has been observed among afebrile infections compared to febrile cases [2, 3]. The histone-modifying Plasmodium homologue of Sir2 (PfSir2a), a transcriptional factor that regulates ribosomal DNA transcription and P. falciparum multiplication rate [4], was found to be more expressed in severe malaria than in uncomplicated infections [5]. Moreover, PfSir2a has been implicated in the epigenetic regulation of var gene expression [6]. This multicopy gene family codes for the P. falciparum erythrocyte membrane protein 1 (PfEMP1) that mediates cytoadhesion of infected erythrocytes to host receptors. var genes can be subdivided as per the motifs present in non-coding sequences and locus position (groups A, B and C) and on the combination of domain cassettes (DCs) [7]. var groups A, B, as well as DC8, 11 and 13 have been associated with severe malaria [7, 8]. Conflicting results have been reported for the associations of var gene expression and asymptomatic malaria [9,10,11], probably due to marked differences in age and parasite densities among febrile and afebrile infections.
Parasite survival, virulence and transmission depend on a balanced investment in within-host replication and between-host transmission [12]. It is hypothesized that the decrease of parasite virulence and increased transmissibility would maximize the long-term persistence of afebrile infections in the human host environment. To test this, parasite transcription of genes involved in cytoadhesion, proliferation and gametocyte maturation were compared between parasites collected from Mozambican individuals with febrile and afebrile outcomes matched by age, parasite density and period of sample collection.
Methods
Study site and population
This study took place in the district of Manhiça (southern Mozambique) where community-based age-stratified cross-sectional surveys were conducted in May of 2012 to 2015. After giving written informed consent, participants were finger-pricked and their blood was used to detect P. falciparum infection by rapid diagnostic test (RDT), microscopy, quantitate real-time PCR (qPCR) and storage in RNAprotect. Plasmodium falciparum-infected individuals were defined as afebrile if their axillary temperature at the time of visit was < 37.5 °C without reporting having had fever in the preceding 24 h, and febrile if the axillary temperature was ≥ 37.5 °C at the time of visit or reported fever during the preceding 24 h. Febrile and afebrile cases were individually matched based on year of sample collection, age (0 to ≤ 5; > 5 to ≤ 15; > 15 to ≤ 25 and > 25 years) and parasite density (≤ 200, > 200 to ≤ 1000, and > 1000 parasites/µL). The Hospital Clínic (Barcelona, Spain) ethics review committee and National Mozambican Ethical Review Committee (Mozambique) approved the study.
Parasitological determinations
Approximately 10 μL of finger-pricked blood were used to perform PfHRP2-based rapid diagnostic tests (SD BIOLINE Malaria Antigen P.f—05FK50) by trained technicians following the manufacturer’s instructions. Thin and thick blood smears were stained with Giemsa as described previously [13]. Two experienced microscopists independently read all slides, and a third reading was performed if there were discrepancies between the results of the first two. Plasmodium falciparum infections were also identified through qPCR from 25 μL of dried blood spots collected on filter papers. DNA was extracted using the QIAamp DNA Mini kit (Qiagen, Hilden, Germany), as per the manufacturer’s instructions. Parasite DNA amplification targeting the 18S rRNA gene was performed using the ABI PRISM 7500 HT Real-Time System (Applied Biosystems, Foster City, USA), following a method described elsewhere [14, 15]. Then, a standard curve was prepared on the foundation of an in vitro culture of the 3D7 strain, which contained known numbers of ring-infected erythrocytes. The standard curve was performed for each test in triplicate with five serial dilutions. Using the 18S rRNA gene as an amplification target, parasitaemia in the clinical samples were quantified and then subjected to extrapolation against the standard curve [16].
RNA extraction
Extraction of RNA from P. falciparum infected blood (50 µL) preserved in 250 µL RNAprotect (Qiagen, Hilden, Germany) was performed using 700 µL of Trizol (Ambion, Life Technology, California, USA) following manual extraction with phenol/chloroform phase separation to elute total RNA. Extracted total RNA was subjected to DNase treatment using DNase Max kit (Qiagen, Hilden, Germany) at 37 °C for 30 min followed by reverse transcription of 400–800 ng of total RNA using PrimeScript™ RT Master Mix reagents (Takara, Shiga, Japan). To confirm the removal of gDNA or the synthesis of cDNA, qPCR was performed using primers targeting P. falciparum ubiquitin-conjugating enzyme (PF08_0085) on reverse transcription positive and negative controls (RNA samples without reverse transcriptase enzyme).
Quantification of transcript levels
Transcript levels of var genes such as varA-exon2, varA-DBLα1 (varA-notDC3), varB group (varB-UTR region) and varC group (varC-UTR region) were assessed using degenerate primers and quantitative reverse transcriptase PCRs (RT-qPCRs) [7, 10, 17]. Domain cassette transcript levels were also determined using a set of primers targeting semi-conserved domains belonging to DC8 (CIDRα1.1), DC11 (CIDRβ2 + DBLγ7) and DC13 (CIDRα1.4) [7, 8]. The assessment of transcript level of genes involved in parasite multiplication (PfSir2a) and gametocyte mature stage (Pfs25) was also performed using previously described RT-qPCR primers [4, 18, 19]. PF08_0085 gene was used as a housekeeping gene [20]. In brief, 96-well plates containing individual 20 μL reactions were amplified in triplicates using a 7500 HT Real-Time System (Applied Biosystem, Foster City, USA). Each reaction mixture had 10 μL of 2X Power SYBR® Green PCR Master Mix (Thermo Fisher Scientific, Warrington, WA, UK), 1 or 0.33 μM of each forward and reverse primers and 5 μL of template cDNA. The reaction volume was prepared with PCR-grade water. Amplifications were performed with a holding for 3 min at 50 °C, initial denaturation for 10 min at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C or 55 °C for 1 min. Samples with a Ct > 40 for any gene target were considered as not expressed. The specificity of primer pairs against human gDNA was also determined. The 7500 System SDS software v1.4 was used to analyse the collected data. A standard curve was then used to calculate the PCR efficiency of each primer from 7 log dilutions of the P. falciparum 3D7 gDNA, by using the formula (E = 10−1/m), where “m” was the slope. The formula C/EΔCt was used to convert Ct values to copy numbers [9]. In the formula, “C” was the number of copies of the gene in the P. falciparum 3D7 genome, “E” was the efficiency of the PCR [7, 9] and ΔCt was the difference in Ct values between a sample and P. falciparum 3D7 reference gDNA loaded in each plate [21]. The relative transcript unit (RTU) of target genes was calculated by dividing the target gene transcript levels by PF08_0085 (housekeeping gene) transcript levels. Non-template controls were tested in every plate [8].
Statistical analysis
Wilcoxon matched-pairs signed rank test and McNemar’s Chi squared test were used to compare continuous and categorical variables, respectively. The association between being febrile or afebrile and log-transformed RTUs was tested by conditional logistic regression models adjusted by area of residence in order to account for differences in transmission intensities between areas. All statistical analyses were performed using R 3.3.2, and GraphPad Prism 5.0 (GraphPad Software) was used to generate graphs.
Results
Among the 3431 participants in cross-sectional surveys from whom blood samples were collected, 278 were P. falciparum positive by qPCR, microscopy and/or RDTs, had information on axillary temperature as well as on fever in the previous 24 h, and had available blood samples in RNAprotect (Fig. 1). Two hundred and one of them (72.3%) were afebrile. Seventy-one of them were matched with febrile individuals with similar age, parasite density and year of sample collection.

Schematic representation of sample selection for reverse transcriptase quantitative PCR analysis
The transcript analysis of target genes was successful in 61 (86.0%) of the 71 pairs (Fig. 1). Thirteen (21.3%) of the febrile individuals had a fever at the time of recruitment and 48 (78.7%) reported having a fever in the preceding 24 h (but had no fever at recruitment). No differences in median age (4.7 years, 95% confidence interval [CI] 2.1–15.2 in afebrile individuals and 5.0 years, 95% CI 3.0–15.1 in febrile individuals, P = 0.979), parasite densities (542.6 parasites/μL of blood, 95% CI 59.7–4267.2 in afebrile individuals and 649.4, 95% CI 45.4–5897.7 in febrile individuals, P = 0.764), sex (P = 0.377) and area of residence (Maragra, Manhiça, Malavele, Palmeria, Taninga and IIha Joshina Machel; P = 0.812) were observed among febrile and afebrile individuals (Table 1).
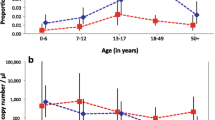
The RT-qPCR efficiencies of targeted genes ranged between 83.0% and 97.6% (Table 2). Among the 9 tested genes, RTUs of 4 of them (varB, varC, DC8 and PfSir2a) were statistically different between afebrile and febrile infections (Fig. 2). The level of varC and PfSir2a transcripts was higher in P. falciparum isolates from afebrile individuals (P ≤ 0.006), while varB and DC8 genes (P ≤ 0.002) were higher in isolates from individuals with febrile infections. After adjusting the analysis by area of residence, doubling the RTU of varC and PfSir2a was associated with a 29.7 (95% CI 4.6–192.3) and 8.5 (95% CI 1.9–32.2) fold increases, respectively, of the odds of being afebrile. In contrast, doubling the RTU of varB and DC8 was associated with a 0.8 (95% CI 0.05–0.6) and 0.2 (95% CI 0.04–0.6) fold changes, respectively, of the odds of being afebrile.

Plasmodium falciparum transcript levels in febrile and afebrile infections. Relative transcript units (RTUs) were calculated with respect to the PF08_0085 housekeeping gene in P. falciparum infections from individuals with fever (axillary temperature ≥ 37.5 °C or reported fever during the preceding 24 h) and afebrile individuals (axillary temperature < 37.5 °C at recruitment) matched by year, age and parasite density. P values were obtained from Wilcoxon matched-pairs signed ranks test. T bars represent median and Interquartile Ranges (IQR). Overall fold change (FC) was calculated as the median and IQR of the RTU ratios between febrile and afebrile pairs
Discussion
This study identified higher abundance of var group C and PfSir2a transcripts in afebrile P. falciparum infections compared to age- and parasite density-matched febrile infections, while opposite patterns were observed for var group B and DC8 transcript levels. Similar gametocyte levels were observed in both types of infections, based on the lack of differences observed for Pfs25 transcripts. Overall, these findings suggest that the expression of variant surface antigens involved in cytoadhesion and immune-evasion can determine the persistence of afebrile infections.
var group B and DC8 genes found to be more expressed in febrile than afebrile infections have been previously linked to cytoadhesion phenotypes (i.e., endothelial protein C receptor) associated with severe malaria [7, 22], thus suggesting a higher pathogenic potential that could drive infections towards clinical manifestations. The higher level of var C transcripts in afebrile infections might indicate the presence of less-pathogenic variant surface antigens compatible with the subclinical persistence of these infections [9]. Increased immunity in afebrile individuals in spite of the matching by host age, or longer durations of infections, may have driven the exhaustion of the parasite var gene repertoire, leading to the expression of var group C variants capable of evading immunity though higher switch-off rates [1, 9]. The higher expression of PfSir2a in afebrile infections might have contributed to this switch towards var C subgroups [6, 23], suggesting the potential involvement of epigenetic mechanisms in afebrile malaria.
This study demonstrates a few noted discrepancies with previous results as well as several limitations. To begin, the association of PfSir2a with afebrile malaria is contradictory to a finding where PfSir2a was found associated with severe malaria [5]. The results of this study may have been influenced by the following limitations (a) febrile individuals identified in the community were not sick enough to seek for clinical care and may present with less pathogenic infections than infections in individuals who attend clinics, (b) uncomplicated and severe malaria patients could co-exist in the febrile group and (c) it is not possible to discard that afebrile infections at the time of the survey may progress to fever during subsequent days. Different criteria have been used to describe afebrile and febrile infections [24, 25]. However, several studies defined infections similarly to the present study [3, 10, 24,25,26,27,28]. Similar gametocyte levels were present in this study. However, higher carriage of gametocyte stages has been observed among afebrile infections at the time of recruitment compared to febrile cases in Tanzania and Burkina Faso [2, 3]. This discrepancy may have been due to matching parasite densities in febrile and afebrile groups of the present study. Moreover, it has been shown that the higher a person’s parasitaemia, the greater the chance of being gametocyte positive [29]. It has also been demonstrated that the detection of Pfs25 transcripts was strongly dependent on parasite density [30]. Lastly, this study targeted a limited number of parasite genes given limited availability of RNA for transcriptional analysis, and may thus have missed some other parasite genes driving afebrile infections.
Conclusion
Major differences exist in var and PfSir2a gene expression between parasites collected from Mozambican individuals with febrile and afebrile outcomes when matched by host age, parasite density and time-period of sample collection. Increased expression of var C genes may increase parasite persistence through a balance between antigenic escape and cytoadhesion to avoid splenic clearance. Several epigenetic mechanisms that include PfSir2a expression might contribute to this transcriptional shift [6]. In contrast, similar gametocyte-specific transcript levels in febrile and afebrile infections suggest that both types of infection are equally transmissible at same parasite density levels.
Availability of data and materials
The datasets analyzed in this study are available from the corresponding author on request.
Abbreviations
- PCR:
-
polymerase chain reaction
- RT-qPCRs:
-
reverse transcriptase quantitative PCRs
- RTU:
-
relative transcript unit
- PfSir2a:
-
Plasmodium homologue of Sir2
- PfEMP1:
-
Plasmodium falciparum erythrocyte membrane protein 1
- DCs:
-
domain cassettes
- RDT:
-
rapid diagnostic test
- qPCR:
-
quantitate real-time PCR
- IQR:
-
interquartile ranges
- FC:
-
fold change
References
Galatas B, Bassat Q, Mayor A. Malaria parasites in the asymptomatic: looking for the hay in the haystack. Trends Parasitol. 2016;32:296–308.
Sumari D, Mwingira F, Selemani M, Mugasa J, Mugittu K, Gwakisa P. Malaria prevalence in asymptomatic and symptomatic children in Kiwangwa, Bagamoyo district, Tanzania. Malar J. 2017;16:222.
Natama HM, Rovira-Vallbona E, Some MA, Zango SH, Sorgho H, Guetens P, et al. Malaria incidence and prevalence during the first year of life in Nanoro, Burkina Faso: a birth-cohort study. Malar J. 2018;17:163.
Mancio-Silva L, Lopez-Rubio JJ, Claes A, Scherf A. Sir2a regulates rDNA transcription and multiplication rate in the human malaria parasite Plasmodium falciparum. Nat Commun. 2013;4:1530.
Merrick CJ, Huttenhower C, Buckee C, Amambua-Ngwa A, Gomez-Escobar N, Walther M, et al. Epigenetic dysregulation of virulence gene expression in severe Plasmodium falciparum malaria. J Infect Dis. 2012;205:1593–600.
Duraisingh MT, Voss TS, Marty AJ, Duffy MF, Good RT, Thompson JK, et al. Heterochromatin silencing and locus repositioning linked to regulation of virulence genes in Plasmodium falciparum. Cell. 2005;121:13–24.
Lavstsen T, Turner L, Saguti F, Magistrado P, Rask TS, Jespersen JS, et al. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proc Natl Acad Sci USA. 2012;109:E1791–800.
Magallon-Tejada A, Machevo S, Cistero P, Lavstsen T, Aide P, Rubio M, et al. Cytoadhesion to gC1qR through Plasmodium falciparum erythrocyte membrane protein 1 in severe malaria. PLoS Pathog. 2016;12:e1006011.
Kaestli M, Cockburn IA, Cortes A, Baea K, Rowe JA, Beck HP. Virulence of malaria is associated with differential expression of Plasmodium falciparum var gene subgroups in a case-control study. J Infect Dis. 2006;193:1567–74.
Rottmann M, Lavstsen T, Mugasa JP, Kaestli M, Jensen AT, Muller D, et al. Differential expression of var gene groups is associated with morbidity caused by Plasmodium falciparum infection in Tanzanian children. Infect Immun. 2006;74:3904–11.
Falk N, Kaestli M, Qi W, Ott M, Baea K, Cortes A, et al. Analysis of Plasmodium falciparum var genes expressed in children from Papua New Guinea. J Infect Dis. 2009;200:347–56.
Rono MK, Nyonda MA, Simam JJ, Ngoi JM, Mok S, Kortok MM, et al. Adaptation of Plasmodium falciparum to its transmission environment. Nat Ecol Evol. 2018;2:377–87.
Gupta H, Matambisso G, Galatas B, Cistero P, Nhamussua L, Simone W, et al. Molecular surveillance of pfhrp2 and pfhrp3 deletions in Plasmodium falciparum isolates from Mozambique. Malar J. 2017;16:416.
Mayor A, Serra-Casas E, Bardaji A, Sanz S, Puyol L, Cistero P, et al. Sub-microscopic infections and long-term recrudescence of Plasmodium falciparum in Mozambican pregnant women. Malar J. 2009;8:9.
Taylor SM, Mayor A, Mombo-Ngoma G, Kenguele HM, Ouedraogo S, Ndam NT, et al. A quality control program within a clinical trial Consortium for PCR protocols to detect Plasmodium species. J Clin Microbiol. 2014;52:2144–9.
Gupta H, Macete E, Bulo H, Salvador C, Warsame M, Carvalho E, et al. Drug-resistant polymorphisms and copy numbers in Plasmodium falciparum, Mozambique, 2015. Emerg Infect Dis. 2018;24:40–8.
Rovira-Vallbona E, Dobano C, Bardaji A, Cistero P, Romagosa C, Serra-Casas E, et al. Transcription of var genes other than var2csa in Plasmodium falciparum parasites infecting Mozambican pregnant women. J Infect Dis. 2011;204:27–35.
Aguilar R, Magallon-Tejada A, Achtman AH, Moraleda C, Joice R, Cistero P, et al. Molecular evidence for the localization of Plasmodium falciparum immature gametocytes in bone marrow. Blood. 2014;123:959–66.
Mancio-Silva L, Zhang Q, Scheidig-Benatar C, Scherf A. Clustering of dispersed ribosomal DNA and its role in gene regulation and chromosome-end associations in malaria parasites. Proc Natl Acad Sci USA. 2010;107:15117–22.
Joice R, Nilsson SK, Montgomery J, Dankwa S, Egan E, Morahan B, et al. Plasmodium falciparum transmission stages accumulate in the human bone marrow. Sci Transl Med. 2014;6:244re245.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45.
Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498:502–5.
Tonkin CJ, Carret CK, Duraisingh MT, Voss TS, Ralph SA, Hommel M, et al. Sir2 paralogues cooperate to regulate virulence genes and antigenic variation in Plasmodium falciparum. PLoS Biol. 2009;7:e84.
Laishram DD, Sutton PL, Nanda N, Sharma VL, Sobti RC, Carlton JM, et al. The complexities of malaria disease manifestations with a focus on asymptomatic malaria. Malar J. 2012;11:29.
Lindblade KA, Steinhardt L, Samuels A, Kachur SP, Slutsker L. The silent threat: asymptomatic parasitemia and malaria transmission. Expert Rev Anti Infect Ther. 2013;11:623–39.
Kiemde F, Tahita MC, Lompo P, Rouamba T, Some AM, Tinto H, et al. Treatable causes of fever among children under five years in a seasonal malaria transmission area in Burkina Faso. Infect Dis Poverty. 2018;7:60.
Magesa SM, Mdira KY, Babiker HA, Alifrangis M, Farnert A, Simonsen PE, et al. Diversity of Plasmodium falciparum clones infecting children living in a holoendemic area in north-eastern Tanzania. Acta Trop. 2002;84:83–92.
Yegorov S, Galiwango RM, Ssemaganda A, Muwanga M, Wesonga I, Miiro G, et al. Low prevalence of laboratory-confirmed malaria in clinically diagnosed adult women from the Wakiso district of Uganda. Malar J. 2016;15:555.
Akim NI, Drakeley C, Kingo T, Simon B, Senkoro K, Sauerwein RW. Dynamics of P. falciparum gametocytemia in symptomatic patients in an area of intense perennial transmission in Tanzania. Am J Trop Med Hyg. 2000;63:199–203.
Nguitragool W, Mueller I, Kumpitak C, Saeseu T, Bantuchai S, Yorsaeng R, et al. Very high carriage of gametocytes in asymptomatic low-density Plasmodium falciparum and P. vivax infections in western Thailand. Parasit Vectors. 2017;10:512.
Acknowledgements
We would like to thank all the study participants, field and laboratory workers at CISM, as well as everyone who supported this study directly or indirectly. We also thank Rebecca Smith Aguasca for her useful comments on this manuscript.
Funding
The study was supported by La Caixa and Bill and Melinda Gates Foundations (OPP1115265) and the Instituto de Salud Carlos III [PI13/01478 cofunded by the Fondo Europeo de Desarrollo Regional (FEDER). AM and HG were further supported by CES10/021-I3SNS and the Department of Science & Technology, Government of India (Overseas Postdoctoral Fellowship, SB/OS/PDF-043/2015-16), respectively. ISGlobal is a member of the CERCA Programme, Generalitat de Catalunya (http://cerca.cat/en/suma/). CISM is supported by the Government of Mozambique and the Spanish Agency for International Development (AECID).
Author information
Authors and Affiliations
Contributions
HG carried out transcript analysis, interpretation of results and wrote the first draft of this manuscript. BG, GM, LN and PC participated in fieldwork, collected clinical and epidemiological data and laboratory analyses. BG, QB, CG, EM, JA, CS, PA, FS and PA participated in the study design, interpretation of results and reviewing of this article. AM coordinated all the stages of the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Hospital Clínic (Barcelona, Spain) ethics review committee and National Mozambican Ethical Review Committee (Mozambique) approved the study. Informed consent and permission (in the case of children under 18 years of age) were also obtained from each participant or a parent/legal guardian during the cross-sectional studies.
Consent for publication
All authors have given their consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gupta, H., Galatas, B., Matambisso, G. et al. Differential expression of var subgroups and PfSir2a genes in afebrile Plasmodium falciparum malaria: a matched case–control study. Malar J 18, 326 (2019). https://doi.org/10.1186/s12936-019-2963-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-019-2963-z