
Abstract
Background
Although Sri Lanka is considered as a malaria-free nation, the threat of re-emergence of outbreaks still remains due to the high prevalence and abundance of malaria vectors. Analysis of population genetic structure of malaria vectors is considered to be one of the vital components in implementing successful vector control programmes. The present study was conducted to determine the population genetic structure of three abundant malaria vectors; Anopheles subpictus sensu lato (s.l.), Anopheles peditaneatus and Anopheles vagus from five administrative districts in two climatic zones; intermediate zone (Badulla and Kurunegala districts) and dry zone (Ampara, Batticoloa and Jaffna districts) of Sri Lanka using the mitochondrial gene, cytochrome c oxidase subunit I (COI).
Methods
Adult mosquitoes of An. subpictus s.l., An. peditaeniatus, and An. vagus were collected from five study sites located in five districts using cattle baited traps and backpack aspirators. Representative samples of each species that were morphologically confirmed were selected from each locality in generating COI sequences (> 6 good quality sequences per species per locality).
Results
Anopheles subpictus s.l. specimens collected during the study belonged to two sibling species; An. subpictus ‘A’ (from all study sites except from Jaffna) and An. subpictus ‘B’ (only from Jaffna). The results of haplotype and nucleotide diversity indices showed that all the three species are having high genetic diversity. Although a high significant pairwise difference was observed between An. subpictus ‘A’ and ‘B’ (Fst> 0.950, p < 0.05), there were no significant genetic population structures within An. peditaeniatus, An. vagus and An. subpictus species A (p > 0.05), indicating possible gene flow between these populations.
Conclusions
Gene flow among the populations of An. peditaeniatus, An. vagus and An. subpictus species A was evident. Application of vector control measures against all mosquito species must be done with close monitoring since gene flow can assist the spread of insecticide resistance genes over a vast geographical area.
Similar content being viewed by others


Background
Knowledge on the population genetic structure of mosquito vectors of disease is vital in understanding their vectorial capacity, in increasing the efficiency of existing vector control programmes and in implementing novel vector control strategies [1,2,3,4,5]. For these reasons, population genetic structures of Anopheles mosquitoes, many species of which are vectors of malaria, have been extensively studied, e.g. Anopheles arabiensis [6, 7], Anopheles baimaii [8], Anopheles culicifacies [9], Anopheles dirus [3, 10, 11], Anopheles funestus [12], Anopheles gambiae [13, 14], Anopheles maculatus [15], Anopheles minimus [16], Anopheles sinensis [17,18,19] and Anopheles stephensi [20, 21].
Studies have shown geographical barriers to be a major determinant of genetic structure of mosquitoes compared to the geographic distance [4, 12, 13, 15, 18]. However, geographic distance and barriers to gene flow can operate in combination to generate population genetic structure e.g. An. sinensis populations in China [17]. Moreover, in Thailand, the genetic structure of Aedes aegypti populations has been shown to be influenced by intense vector control activities [5].
Several mitochondrial DNA (mtDNA) regions have been used as successful genetic markers in barcoding of mosquitoes and, in analyzing the genetic diversity and genetic structure of populations. Among these markers, cytochrome c oxidase subunit I (COI) has been the most extensively used marker in studies on the genetic structure of mosquitoes, including An. sinensis [17], An. baimaii [8], An. dirus [10, 11, 22], An. lesteri [23], An. darling [24], An. stephensi [21] and Aedes albopictus [25].
The mosquito fauna of Sri Lanka is represented by 141 species, of which 23 belong to the genus Anopheles. Species An. culicifacies and An. subpictus are considered respectively as primary and secondary vectors of malaria [26,27,28]. Whereas Anopheles aconitus, Anopheles annularis, Anopheles barbirostris, Anopheles nigerrimus, Anopheles pallidus, Anopheles peditaneatus, Anopheles tessellatus, Anopheles vagus and Anopheles varuna are considered as potential malaria vectors in Sri Lanka [29, 30]. Also An. stephensi, one of the major malaria vectors in India was recently discovered from northwestern coasts of Mannar in Sri Lanka [31, 32]. Although the World Health Organization declared Sri Lanka a malaria-free nation in 2016, there is a high risk of reemergence of the disease with an introduction of the parasite, especially through travelers from malaria endemic countries, as the vectors are available throughout the country [33]. Currently the country keeps the vectors suppressed mainly through the use of a combination of organophosphates and pyrethroids in vector control programmes.
Continuous exposure to insecticides over a long period of timed imposes a great selection pressure to develop insecticide resistance in mosquito populations. Both the major vectors An. culicifacies and An. subpictus, and several other potential vector species including An. peditaeniatus and An. nigerrimus have developed resistance to a range of insecticides from all the major groups; organochlorines, organophosphates, carbamates and pyrethroids [34]. It has been shown that the gene flow play an important role in the spread of resistance genes in mosquito populations [35,36,37]. Therefore, resistance genes developed in a vector population of one particular area can be spread effectively into other areas of the country through the gene flow.
Among the malaria vectors found in Sri Lanka, An. culicifacies, An. subpictus, An. annularis and An. barbirostris occur as species complexes [33]. Anopheles subpictus exists as a sibling species complex and studies have shown the occurrence of two genetically distinct entities of this species; An. subpictus ‘A’ and An. subpictus ‘B’ [26, 33]. Of the two members of Culicifacies complex present in Sri Lanka, An. culicifacies species E is the vector of malaria parasite whereas B is a non-vector. Species E always has shown relatively high resistance to commonly used insecticides than species B [9]. Population genetic structure analysis of An. culicifacies E using microsatellite data has shown the effect of geographic barriers on the genetic variation of this species [38]. As sibling species can have different feeding habits, behavior patterns, disease transmission rates, similar control measures might not be effective against different sibling species.
Hence, studies on genetic diversity and population structure of malaria vectors is important in implementing successful vector control programmes against the reemergence of malaria in the country. Few studies have been carried out to determine the population genetic structure of Sri Lankan An. culicifacies previously [9, 38]. This study aims to analyse the population genetic structure of another three important malaria vectors An. subpictus, An. peditaneatus, and An. vagus using the mitochondrial gene, cytochrome c oxidase subunit I (COI), for the first time in Sri Lanka.
Methods
Study sites and mosquito collection
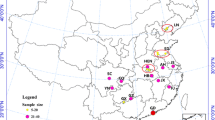
Mosquitoes were collected from five districts of Sri Lanka. A single locality was selected from each district; Kalmunai in Ampara district, Haldummulla in Badulla district, Batticaloa in Batticaloa district, Tirunelveli in Jaffna district, Wariyapola in Kurunegala district (Fig. 1). These sites are located in the following climatic zones in Sri Lanka i.e. Haldummulla in up country intermediate zone (> 900 m elevation 1750–2500 mm rainfall); Wariyapola in low country intermediate zone (0–300 m elevation, 1750–2500 mm rainfall); Kalmunai, Batticaloa and Tirunelveli in low country dry zone (0–300 m elevation, < 1750 mm rainfall with a distinct dry period) (Fig. 1). The highest geographic distance was between Tirunelveli and Kalmunai (322 km) and lowest was between Batticaloa and Kalmunai (37 km) study sites.

Five study sites located in each district where An. peditaeniatus, An. subpictus s.l. and An. vagus mosquitoes were collected for the population genetic structure analysis (elevations of the study sites are given in parentheses; green—intermediate zone, grey—dry zone)
Adult mosquitoes were collected monthly using cattle baited traps (one trap in each study site) and backpack aspirators from January 2014 to July 2015. Samples were collected from 2 to 3 points from each study site. These localities were selected based on previous study results where high abundance of these vectors was reported in all five selected study sites [33]. Dried specimens were morphologically identified into species level using standard taxonomic keys [39] and stored for molecular characterization. A representative randomly selected samples of each species from each locality was selected for sequencing.
DNA extraction, polymerase chain reaction (PCR) and sequencing
Genomic DNA was extracted from head and thoracic regions of each morphologically identified individuals using nexttec™ DNA Isolation Kits (Nexttec Biotechnologies GmbH, Leverkusen, Germany), according to the manufacturer’s protocol.
A region of the COI gene was amplified using forward primer C1-J-1718 (5′-GGAG GATTTGGAAATTGATTAGTTCC-3′) and reverse primer C1-N-2191 (5′ CCCGGTAAAATTAAAATATAAACTTC-3′) [40]. Each amplification was performed in 15 µl that included 1 µl of DNA template, 1.5 µl of 10× KAPA buffer A, 0.12 µl of KAPA taq, 0.12 µl of 2.5 mM dNTP mix, 0.75 µl of 50 mM MgCl2, 0.51 µl of each primer (10 mmol) and 10.49 µl of ddH2O. The PCR parameters were 95 °C for 5 min and 35 cycles of 94 °C for 30 s, 51 °C and 72 °C for 45 s, followed by a final extension step of 72 °C for 10 min. PCR products were run in 1.5% agarose gel stained with Medori green and visualized in a gel imaging system.
PCR products showing positive clear bands were purified using QIAquick® PCR Purification kits according to the manufacturers’ protocol. A minimum of six PCR positive samples of each species from each district were sequenced bidirectionally at Source Bioscience, Nottingham, United Kingdom.
Statistical analysis
The trace files/chromatograms of COI sequences (a minimum of 6 sequences for each species from each district) were manually edited using BioEdit software. Sequences of low quality were excluded and a minimum of 6 good quality sequences from each species from each locality were used for data analysis. After trimming the COI sequences to remove ambiguous sites, final fragments of 403 bp in An. peditaeniatus and An. subpictus and, 423 bp in An. vagus were used in the genetic diversity and population genetic structure analysis. Once the alignment was completed, sequences were compared with the publicly available sequence data in GenBank using BLAST [41] and the BOLD interface [42] to confirm species identification. Amino acid sequences were inferred to check for the presence of stop codons. Number of haplotypes (h), genetic diversity indices [Haplotype Diversity Index (Hd) and Nucleotide Diversity Index (Pi)] and, Neutrality tests (Tajima’s D and Fu’s Fs) were performed in DNA Sequences Polymorphism software (dnaSP) (version 5.1.10). Pairwise differences and population structures of each species were evaluated by analysis of molecular variance (AMOVA) in Arlequin 3.11 and significance was evaluated based on 10,000 permutations. Based on the number of nucleotide differences, haplotype networks of these three species were constructed using Network software 5.0.0.1 to determine the interrelationship between haplotypes.
Results
Translated amino acid sequences revealed that there are no frame shifts or stop codons in all the edited sequences, indicating the mitochondrial origin of the DNA. Comparison of COI sequences of An. peditaeniatus and An. vagus with the publicly available sequences completely agreed with our morphological identification. The morphologically identified An. subpictus s.l., specimens belonged to two genetic entities. All the specimens from Jaffna belonged to An. subpictus species B while specimens from the other four sites belonged to An. subpictus species A. The haplotype diversities (Hd) and nucleotide diversities (Pi) were similarly high for all species except for An. subpictus species B which reported relatively low Hd (0.666) and Pi (0.002) (Table 1). According to neutrality test results, both Tajima’s D and Fu’s Fs values were not significant in any of the species (p > 0.100) (Table 1).
Anopheles peditaeniatus showed the highest number of haplotype sharing among the five studied localities (4 shared haplotypes) followed by An. subpictus species A (3 shared haplotypes) and An. vagus (1 shared haplotype). Anopheles subpictus species B was present only in the Jaffna study site. The most dominant haplotype of An. peditaeniatus (33.33% of the total number of haplotypes) and An. vagus (35.48% of the total number of haplotypes) were shared among 4 localities while that of An. subpictus species A (25.71% of the total number of haplotypes) was shared only between three localities as shown in Fig. 2.

Haplotype networks generated using Network 5.0.0.1 for a An. peditaeniatus, b An. vagus and c An. subpictus s.l. collected from five geographical locations in Sri Lanka. Each haplotype is represented by a circle and the size of the circle is proportional to the number of individuals with each haplotype. Geographical localities are colour coded
Unlike the haplotype network drawn for An. peditaeniatus and An. vagus the haplotype network of An. subpictus s.l. formed two distinct clusters; one representing 16 haplotypes of An. subpictus species A and the other representing 2 haplotypes of An. subpictus species B (Fig. 2c).
The pairwise comparison of population differentiation is presented in Table 2. Anopheles subpictus species B population from Jaffna showed a very high significant pairwise difference with four An. subpictus species A populations with FST values always greater than 0.950 (p < 0.05) (Table 2). Population pairwise FST values within An. peditaeniatus, An. subpictus species A and An. vagus were not significant indicating an absence of genetic differentiation among populations within these species (Table 2).
AMOVA was conducted to estimate the genetic structure variation among populations of each species and the results obtained are shown in Additional file 1. According to the variations estimated for An. peditaeniatus, An. vagus, and An. subpictus species A, a significant variation was observed among individuals within populations (percentage variation; 101.99% for An. peditaeniatus and 102.29% for An. vagus and 100.47% for An. subpictus species A) (p < 0.05).
Discussion
Suppression of the malaria vector population is the most effective way of preventing the re-emergence of malaria outbreaks in Sri Lanka. The present study was conducted to analyze population genetic structure of three abundant malaria vectors, as this knowledge is important in planning future vector control programmes of Sri Lanka.
The present study also reports the presence of two genetic entities of An. subpictus; “species A” and “species B” confirming the results of the previous study on barcoding of Anopheline mosquitoes from the same study sites [33]. High Fst values obtained during the comparison between An. subpictus species A and species B populations, indicated that these two are genetically distinct from each other.
Both pairwise comparisons (Fst values) and the analysis of molecular variance (AMOVA) showed that there is no genetic structure variation in An. peditaeniatus, An. vagus and An. subpictus species A populations used during the current study. These species showed haplotype sharing between the five populations and it was highest for An. peditaeniatus. A mechanism of mixing of these mosquito populations from different geographical areas and possible gene flow is evident by these observations even though a considerably high geographic distance, ranging from 32 to 322 km, is present between these study sites. Sri Lanka is an island with 103 rivers basins and all these three species of mosquitoes breed in variety of freshwater habitats, which are connected to these riverine systems one way or the other. Further, all these rivers start from the mountainous areas at the center of the country and flow through the three climatic zones (wet, intermediate and dry zone) before joining the sea. Hence, the sites of the current study are connected by mosquito habitats, which allow gene flow between these localities. Further, the rainfall experienced by these study sites also build up this connection between the breeding sites.
Sri Lanka is an island with a relatively small land area and there were no major geographical barriers between the studied localities. Hence, regardless of the geographic distance the possibility of gene flow between the study sites is considerably higher. Anopheles maculatus populations that have been separated by 50 km have shown limited gene flow in the presence of geographic barriers while in the absence of geographic barriers free gene flow has been observed even between populations 650 km apart [15]. Several other studies related to mosquitoes have also reported absence of correlation between genetic isolation and geographic distance [4, 5, 7, 18, 19].
However, a study using COI marker and microsatellites has shown that the geographic distance has an effect on the genetic structure variation of Sri Lankan An. culicifacies populations [9]. Although the central mountain range of Sri Lanka has acted as a barrier for the gene flow of An. culicifacies E, it was not a barrier for An. peditaeniatus, An. vagus and An. subpictus species A [38]. Therefore, it appears that the relationship between geographic distance and the population genetic structure of anophelines depends on the type of the species probably due to species wise variations in breeding habitats, breeding patterns and behaviour.
Continuity of mosquito breeding sites supported by the absence of geographical barriers can be considered as the main reason for the maintenance of gene flow between the An. peditaeniatus, An. vagus and An. subpictus species A populations in Sri Lanka. Regular monitoring of population genetic structure of malaria vectors is important in developing effective vector control strategies to address the possible impact made by the spread of vital genes such as insecticide resistance genes through vector populations.
Conclusions
Anopheles subpictus s.l. collected from five Sri Lankan districts belonged to two genetically distinct species An. subpictus species A and An. subpictus species B. Gene flow was evident even between geographically distant populations of An. peditaeniatus, An. vagus and An. subpictus species A perhaps due to absence of geographic barriers and the continuity of habitats. Results also validated the use of COI gene as a tool in understanding gene flow of anophelines in Sri Lanka.
Abbreviations
- COI :
-
Cytochrome c oxidase subunit I
- mtDNA:
-
mitochondrial DNA
- Hd:
-
Haplotype Diversity Index
- Pi:
-
Nucleotide Diversity Index
- dnaSP:
-
DNA Sequences Polymorphism software
- AMOVA:
-
analysis of molecular variance
References
Chen H, Minakawa N, Beier J, Yan G. Population genetic structure of Anopheles gambiae mosquitoes on Lake Victoria islands, west Kenya. Malar J. 2012;3:48.
Donnelly MJ, Simard F, Lehmann T. Evolutionary studies of malaria vectors. Trends Parasitol. 2002;18:75–80.
Walton C, Handley JM, Collins FH, Baimai V, Harbach R, Deesin V, et al. Genetic population structure and introgression in Anopheles dirus mosquitoes in Southeast Asia. Mol Ecol. 2001;10:569–80.
Olanratmanee P, Kittayapong P, Chansang C, Hoffmann AA, Weeks AR, Endersby NM. Population genetic structure of Aedes (Stegomyia) aegypti (L.) at a micro-spatial scale in Thailand: implications for a dengue suppression strategy. PLoS Negl Trop Dis. 2013;7:e1913.
Gorrochotegui-Escalante N, Gomez-Machorro C, Lozano-Fuentes S, Fernandez-Salas L, De Lourdes-Muñoz ML, Farfan-Ale JA, et al. The breeding structure of Aedes aegypti populations in Mexico varies by region. Am J Trop Med Hyg. 2002;66:213–22.
Muturi EJ, Kim C, Baliraine FN, Musani S, Jacob B, Githure J, Novak RJ. Population genetic structure of Anopheles arabiensis (Diptera:Culicidae) in a rice growing area of central Kenya. J Med Entomol. 2010;47:144–51.
Nyanjom SRG, Chen H, Gebre-Michael T, Bekele E, Shililu J, Githure J, et al. Population genetic structure of Anopheles arabiensis mosquitoes in Ethiopia and Eritrea. J Hered. 2003;94:457–63.
Sarma DK, Prakash A, O’Loughlin SM, Bhattacharyya DR, Mohapatra PK, Bhattacharjee K, et al. Genetic population structure of the malaria vector Anopheles baimaii in north-east India using mitochondrial DNA. Malar J. 2012;11:76.
Surendran SN, Truelove N, Sarma DK, Jude PJ, Ramasamy R, Gajapathy K, et al. Karyotypic assignment of Sri Lankan Anopheles culicifacies species B and E does not correlate with cytochrome oxidase subunit I and microsatellite genotypes. Parasit Vectors. 2015;8:327.
O’Loughlin SM, Okabayashi T, Honda M, Kitazoe Y, Kishino H, Somboon P, et al. Complex population history of two Anopheles dirus mosquito species in Southeast Asia suggests the influence of Pleistocene climate change rather than human-mediated effects. J Evol Biol. 2008;21:1555–69.
Walton C, Handley JM, Tun-Lin Collins FH, Harbach RE, Baimai V, et al. Population structure and population history of Anopheles dirus mosquitoes in Southeast Asia. Mol Biol Evol. 2000;17:962–74.
Braginets OP, Minakawa N, Mbogo CM, Yan G. Population genetic structure of the African malaria mosquito Anopheles funestus in Kenya. Am J Trop Med Hyg. 2003;69:303–8.
Markianos K, Bischoff E, Mitri C, Guelbeogo WM, Gneme A, Eiglmeier K, et al. Genetic structure of a local population of the Anopheles gambiae complex in Burkina Faso. PLoS One. 2016;11:e0145308.
Lehmann T, Hawley WA, Kamau L, Fontenille D, Simard F, Collins FH. Genetic differentiation of Anopheles gambiae populations from East and West Africa: comparison of microsatellite and allozyme loci. J Hered. 1996;77:192–208.
Rongnoparut P, Rodpradit P, Kongsawadworakul P, Sithiprasasna R, Linthicum KJ. Population genetic structure of Anopheles maculatus in Thailand. J Am Mosq Control Assoc. 2006;22:192–7.
Van Bortel W, Trung HD, Roelants P, Backeljau T, Coosemans M. Population genetic structure of the malaria vector Anopheles minimus A in Vietnam. Heredity. 2003;91:487–93.
Feng X, Huang L, Lin L, Yang M, Ma Y. Genetic diversity and population structure of the primary malaria vector Anopheles sinensis (Diptera:Culicidae) in China inferred by COI gene. Parasit Vectors. 2017;10:75.
Ma Y, Yang M, Fan Y, Wu J, Xu J. Population structure of the malaria vector Anopheles sinensis (Diptera:Culicidae) in China: two gene pools inferred by microsatellites. PLoS One. 2011;67:e22219.
Ma Y, Qu F, Xu J, Zheng Z. Study on molecular genetic polymorphism of Anopheles sinensis populations in China. Acta Entomol Sin. 2001;44:33–9.
Gakhar SK, Sharma R, Sharma A. Population genetic structure of malaria vector Anopheles stephensi Liston (Diptera:Culicidae). Indian J Exp Biol. 2013;51:273–9.
Ali N, Hume JC, Dadzie SK, Donnelly MJ. Molecular genetic studies of Anopheles stephensi in Pakistan. Med Vet Entomol. 2007;21:265–9.
Wang D, Ma Y, Zhou H. Genetic variation of Anopheles dirus A and D (Diptera:Culicidae) in China: inferred by mtDNA-COI gene sequences. Chinese Journal of Parasitology and Parasitic Diseases. 2007;25:368–71 (in Chinese).
Yang M, Ma Y. Molecular population genetic structure of Anopheles lesteri (Diptera:Culicidae) based on mtDNA-COI gene sequences. Acta Entomol Sin. 2009;52:1000–7.
Gutierrez LA, Gomez GF, Gonzalez JJ, Castro MI, Luckhart S, Conn JE, et al. Microgeographic genetic variation of the malaria vector Anopheles darling Root (Diptera:Culicidae) from Cordoba and Antioquia, Colombia. Am J Trop Med Hyg. 2010;83:38–47.
Zawani MKN, Abu HA, Sazaly AB, Zary SY, Darlina MN. Population genetic structure of Aedes albopictus in Penang, Malaysia. Genet Mol Res. 2014;13:8184–96.
Surendran NS, Sarma DK, Jude PJ, Kemppainen P, Kanthakumaran N, Gajapathy K, et al. Molecular characterization and identification of members of the Anopheles subpictus complex in Sri Lanka. Malar J. 2013;12:304.
Gunathilaka N, Karunaraj P. Identification of sibling species status of Anopheles culicifacies breeding in polluted water bodies in Trincomalee district of Sri Lanka. Malar J. 2015;14:214.
Gunathilaka N. Illustrated key to the adult female Anopheles (Diptera:Culicidae) mosquitoes of Sri Lanka. Appl Entomol Zool. 2017;52:69–77.
Amerasinghe PH, Amerasinghe FP, Konradsen F, Fonseka KT, Wirtz RA. Malaria vectors in a traditional dry zone village in Sri Lanka. Am J Trop Med Hyg. 1999;60:421–9.
Herath PRJ, Abeywardena T, Padmalal UK. A study of the role of different indigenous anopheline species in the transmission of human malaria in Sri Lanka. Proc Sri Lanka Assoc Adv Sci. 1983;39:6.
Dharmasiri AGG, Perera AY, Harishchandra J, Herath H, Aravindan K, Jayasooriya HRT, et al. First record of Anopheles stephensi in Sri Lanka: a potential challenge for prevention of malaria reintroduction. Malar J. 2017;16:326.
Surendran SN, Sivabalakrishnan K, Gajapathy K, Arthiyan S, Jayadas TTP, Karvannan K, et al. Genotype and biotype of invasive Anopheles stephensi in Mannar Island of Sri Lanka. Parasit Vectors. 2018;11:3.
Weeraratne TC, Surendran SN, Reimer LJ, Wondji CS, Perera MDB, Walton C, et al. Molecular characterization of Anopheline (Diptera:Culicidae) mosquitoes from eight geographical locations of Sri Lanka. Malar J. 2017;16:234.
Perera MDB, Hemingway J, Karunaratne SHPP. Multiple insecticide resistance mechanisms involving metabolic changes and insensitive target sites selected in anopheline vectors of malaria in Sri Lanka. Malar J. 2008;7:168.
Barnes KG, Irving H, Chiumia M, Mzilahowa T, Coleman M, Hemingway J, et al. Restriction to gene flow is associated with changes in the molecular basis of pyrethroid resistance in the malaria vector Anopheles funestus. Proc Natl Acad Sci USA. 2017;114:286–91.
Pasteur N, Raymond M. Insecticide resistance genes in mosquitoes: their mutations, migration, and selection in field populations. J Hered. 1996;87:444–9.
Raymond M, Berticat C, Weill M, Pasteur N, Chevillon C. Insecticide resistance in the mosquito Culex pipiens: what have we learned about adaptation? Genetica. 2001;112:287–96.
Harischandra IN, Dassanayake RS, De Silva BG. Three sympatric clusters of the malaria vector Anopheles culicifacies E (Diptera:Culicidae) detected in Sri Lanka. Parasit Vectors. 2016;9:3.
Amerasinghe FP. A Guide to the identification of the Anopheline mosquitoes (Diptera:Culicidae) of Sri Lanka. I. Adult females. Ceylon J Sci. 1990;21:1–16.
Simon C, Frati F, Beckenbach A, Crepsi B, Liu H, Flook K. Evolution, weighting and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994;87:651–701.
NCBI National Center for Biotechnology. Information. http://www.blast.ncbi.nlm.nih.gov. Accessed 20 Feb 2016.
BOLDSYSTEMS. http://www.boldsystems.org. Accessed 5 Mar 2016.
Authors’ contributions
SHPPK conceptualized and designed the work. TCW conducted morphological identifications of mosquitoes and molecular laboratory work. TCW, CW, SNS and SHPPK analysed the data. All contributed to the discussion and writing the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Field assistance by Dr. DevikaPerera (Regional Malaria officer, Anti-Malaria Campaign, Kurunegala, Sri Lanka) is greatly acknowledged.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets generated during the current study are available in the GenBank repository, (http://www.ncbi.nlm.nih.gov/genbank/). All data generated during this study are included in this published article (Additional file 1).
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
International Research Center (InRC), University of Peradeniya, Sri Lanka (Grant No. InRC/RG/13/21) for funding in conducting field work and laboratory work in Sri Lanka Royal Society, UKTravel Grant to SHPPK to work with University of Manchester (Commonwealth Science Conference Follow-on Grants 2015).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1.
Results of COI genetic structure variations estimated using AMOVA for An. subpictus, An. peditaeniatus and An. vagus collected from five geographical locations in Sri Lanka.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Weeraratne, T.C., Surendran, S.N., Walton, C. et al. Genetic diversity and population structure of malaria vector mosquitoes Anopheles subpictus, Anopheles peditaeniatus, and Anopheles vagus in five districts of Sri Lanka. Malar J 17, 271 (2018). https://doi.org/10.1186/s12936-018-2419-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-018-2419-x