
Abstract
Background
Primaquine (PQ) is recommended as an addition to standard malaria treatments in pre-elimination settings due to its pronounced activity against mature Plasmodium falciparum gametocytes, the parasite stage responsible for onward transmission to mosquitoes. However, PQ may trigger haemolysis in glucose-6-phosphate dehydrogenase (G6PD)-deficient individuals. Additional human genetic factors, including polymorphisms in the human cytochrome P450 2D6 (CYP2D6) complex, may negatively influence the efficacy of PQ. This study assessed the prevalence of G6PD deficiency and two important CYP2D6 variants in representative pre-elimination settings in South Africa, to inform malaria elimination strategies.
Methods
Volunteers (n = 248) attending six primary health care facilities in a malaria-endemic region of South Africa were enrolled between October and November 2015. G6PD status was determined phenotypically, using a CareStart™ G6PD rapid diagnostic test (RDT), and genotypically for two common African G6PD variants, namely A+ (A376G) and A− (G202A, A542T, G680T & T968C) by PCR, restriction fragment length polymorphisms (RFLP) and DNA sequencing. CYP2D6*4 and CYP2D6*17 variants were determined with PCR and RFLP.
Results
A prevalence of 13% (33/248) G6PD deficiency was observed in the cohort by G6PD RDT whilst by genotypic assessment, 32% (79/248) were A+ and 3.2% were A−, respectively. Among the male participants, 11% (6/55) were G6PD A− hemizygous; among females 1% (2/193) were G6PD A− homozygous and 16% (32/193) G6PD A− heterozygous. The strength of agreement between phenotyping and genotyping result was fair (Cohens Kappa κ = 0.310). The negative predictive value for the G6PD RDT for detecting hemizygous, homozygous and heterozygous individuals was 0.88 (95% CI 0.85–0.91), compared to the more sensitive genotyping. The CYP2D6*4 allele frequencies for CYP2D6*4 (inferred poor metabolizer phenotype) and CYP2D6*17 (inferred intermediate metabolizer phenotype) were 3.2 and 19.5%, respectively.
Conclusions
Phenotypic and genotypic analyses both detected low prevalence of G6PD deficiency and the CYP2D6*4 variants. These findings, combined with increasing data confirming safety of single low-dose PQ in individuals with African variants of G6PD deficiency, supports the deployment of single low-dose PQ as a gametocytocidal drug. PQ would pose minimal risks to the study populations and could be a useful elimination strategy in the study area.
Similar content being viewed by others

Background
South Africa is one of eight southern African countries that are signatories of the Elimination 8 (E8) initiative, a multi-country coordinated effort to achieve malaria elimination. The four frontline countries (South Africa, Botswana, Namibia and Swaziland) aim for elimination, in various timeframes by 2020 [1], thus setting the stage for subsequent elimination in the other E8 member states [2]. In an effort to accelerate the region towards malaria elimination, novel parasite and integrated vector tools are being explored.
For treatment of patients with Plasmodium falciparum malaria, the World Health Organization (WHO) advocates the use of a single low-dose (0.25 mg/kg) of primaquine (PQ) together with artemisinin-based combination therapy in malaria-eliminating countries [3]. PQ is effective against the transmissible gametocyte stage of P. falciparum parasites and may reduce onward transmission to mosquitoes if deployed at community level [4, 5]. It is also the only commercially-available drug active against hypnozoites in Plasmodium vivax and Plasmodium ovale infections [6]. However, country-specific efforts towards low-dose PQ deployment for P. falciparum transmission reduction are being impeded by the perceived safety issue surrounding PQ use in glucose-6-phosphate dehydrogenase (G6PD)-deficient individuals. When exposed to high doses of 8-aminoquinoline anti-malarial agents like PQ and tafenoquine, G6PD-deficient individuals risk suffering haemolysis [6, 7]. However, at the recommended low dose, the risk of haemolysis is thought to be substantially reduced and PQ is considered safe [3].
G6PD deficiency is an X-linked genetic disorder that displays great polymorphism with about 186 described mutations [8], resulting in the most common enzymopathy in the world [7]. G6PD deficiency is clinically observed mainly in hemizygous males or homozygous females, with heterozygous females presenting with a wide spectrum of aberrant enzyme activities due to mosaicism. Circumstantial evidence suggests that G6PD deficiency imparts some resistance against malaria as evidenced by the overlap in the geographical distribution of the deficiency with present and past malaria endemicity [9], although the exact nature of such potential protection is unknown. Whilst earlier studies reported G6PD deficiency protection against severe forms of malaria [10, 11], recent data from a large multi-centre case control study and a meta-analysis suggests protection from cerebral malaria and high parasitaemia and not severe malarial anaemia, mainly in heterozygous individuals in African settings [12, 13]. Although the complexity of this disorder presents a diagnostic challenge, certain G6PD variants, including the African and Mediterranean variants, have been used to probe the general G6PD deficiency status in populations [14]. G6PD variants are classified according to their residual enzyme activity into three types [15]. These range from the most severe presentations but rare type 1, that present with congenital nonspherocytic anaemia, while type 2 are usually asymptomatic in the steady state until exposed to exogenous triggers. These type 2 variants are the equivalent of class II and III of the WHO classification system [8] and predisposes individuals to substantial risk of haemolytic anaemia. Type 2 variants include the Mediterranean, A−, Mahidol, Canton, Vanua Lava and Seattle variants amongst others [16]. Type 3 variants manifest phenotypically as normal G6PD activity with no clinical issues.
In sub-Saharan Africa, three variants are most commonly described, namely the B wild-type variant associated typically with normal G6PD activity and the G6PD African variants, A+ and A− [16]. The African variants (“A”) are amongst the most common in the world with a wide enzyme activity range [7]. The A+ variant carries an A376G mutation in exon V which results in mild enzyme deficiencies. By contrast, the more common A− variant which carries an additional mutation in exon IV (G202A) is associated mild clinical presentations such as jaundice and dark urine when exposed to 8-aminoquinolines [16]. Several other A− mutations (A542T, G680T and T968C) have been detected in West African individuals [17, 18].
PQ efficacy is further modulated by the human cytochrome P450 2D6 (CYP2D6) enzyme [19], which metabolizes ~ 25% of all clinically prescribed drugs [20]. CYP2D6 is highly polymorphic with varied enzymatic activity as a result of allelic sequence variations and protein structural rearrangements. These genotypic and phenotypic variations affect drug metabolism, probably increasing the risk of treatment failure or potential toxicity [21]. To date, more than 113 allelic variants (and sub-variants) of the CYP2D6 gene have been reported [22], which can be classified in four distinct phenotypic groups. Poor metabolizers (PMs) have two copies of defective alleles resulting in reduced expression of a particular CYP2D6 enzyme. Intermediate metabolizers (IMs) are heterozygous for a defective allele and a functional allele resulting in slightly reduced enzyme activity. Extensive metabolizers (EMs) carry two functional alleles and have normal enzyme activity, while ultra-rapid metabolizers (UMs) have more than two functional gene copies and elevated enzyme activity [23]. The PMs with the CYP2D6*4 polymorphism are associated with PQ treatment failure in P. vivax [24], whereas IMs harbouring CYP2D6*17 mutations have been linked with diminished enzyme activity in black Africans [25]. Whilst the impact of CYP2D6 metabolizer status on the efficacy of single low-dose PQ for gametocyte clearance is currently unknown, it is conceivable that the gametocytocidal effect of PQ would be reduced if lower plasma concentrations of the active PQ metabolite are achieved [26].
Prevalence of G6PD deficiency and CYP2D6 polymorphisms in a population are thus important considerations for public health interventions involving the deployment of 8-aminoquinoline drugs like PQ [27]. In sub-Saharan Africa, prevalence of G6PD deficiency is estimated to range between 5 and 37.5% [9], and CYP2D6 allele frequencies range from 1 to 33% [28]. South Africa has previously (1940s–1980s) reported fewer cases of G6PD deficiency (5.3%) [29, 30] and CYP2D6 (2.6%) [31] mutations compared to its neighbouring countries, most likely a consequence of genetic differences among the distinct ethnic groups [32, 33]. However, recent population movements and immigration [34] may have led to shifts in the frequency of the enzyme deficiency and CYP2D6 variants across the country. Due to a paucity of current data, regionally-relevant evidence of G6PD deficiency and CYP2D6 variant metrics are needed. These will inform implementation of single low-dose PQ policy in pre-elimination settings within South Africa.
This study aimed to determine the current prevalence of both G6PD deficiency and CYP2D6 variants in representative populations in a malaria-endemic region of South Africa. G6PD deficiency measurements were also compared to phenotypic screening with a potential point-of-care tool in mass screen and test scenarios. This is the first study employing genotyping to evaluate locally relevant G6PD deficiency and CYP2D6 variants in a pre-elimination setting of South Africa.
Methods
Ethics statement
Ethical approval for the study was obtained from the ethical review committees of the University of Pretoria (Ethics reference No. 406/2014) and the Limpopo Department of Health (Ref 4/2/2). Signed informed consent in the local language (Tshivenda) was obtained from all participants prior to sample collection. Parents or guardians of children less than 7 years of age gave written, informed consent, while children between the ages of 7 and 17 years gave assent before sampling. An adult witness was required to co-sign the informed consent document.
Study site and population
This study was conducted in the Vhembe District, a 25,597 km2 area in Limpopo Province, a malaria-endemic region in the northernmost part of South Africa. Malaria transmission primarily occurs during the months of September to May, with limited transmission generally occurring over the winter months of June and July [35]. Vhembe District is responsible for more than 60% of all reported malaria cases in South Africa [36], with 1543 cases and 18 deaths noted in the region in the 2015/16 transmission season, when sampling occurred [37, 38]. The district is bordered by Mozambique to the south-east, Zimbabwe to the north and Botswana to north-west. The climate is typically subtropical with mild winters and wet, warm summers with average rainfall per annum of 820 mm [39]. The district experiences frequent droughts with areas that are predominantly semi-arid [40].
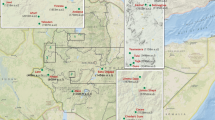
Study participants were recruited from six primary health care clinics (PHCs): Folovhodwe, Madimbo, Manenzhe, Masisi, Mulala and Tshipise in Mutale and Musina local municipalities (Fig. 1) during October and November 2015. Once recruited into the study in one particular clinic, patients were ineligible for subsequent enrolment even if they presented at a different clinic. The district has an estimated population of 1,347,235 with a population density of 52.6 persons per km2 [41]. The area is mainly inhabited by the Venda ethnic group, with sizable Zimbabwean immigrant populations (Shona and Ndebele ethnic groups) that work on the commercial farms and reside in the area.

Map of the study site in Vhembe District, Limpopo Province, South Africa. The six primary health care clinics (H) and village catchment areas where participants were recruited are indicated. The study district in Limpopo Province is shown alongside other malaria-endemic provinces of KwaZulu-Natal and Mpumalanga, is in close proximity to Botswana, Zimbabwe and Mozambique
Questionnaire administration and sample collection
A brief questionnaire on demographics (specifically age and gender), travel history, history of fever or any malaria symptoms, ethnicity and residence was administered to the consenting participants. Upon completion of the questionnaire, finger prick filter paper (Munktell TFN card, Lasec, Cape Town, South Africa) blood spots were collected from the participants for P. falciparum parasite detection and human genotyping studies. Sample size in this pilot study was constrained by the availability of CareStart™ G6PDd rapid diagnostic test (RDT) kits.
Malaria parasite detection
Peripheral finger prick blood samples were tested for P. falciparum parasite antigens using RDT kits detecting the histidine-rich protein 2 (First Response® Malaria Antigen P. falciparum card test HRP2, Premier Medical Corporation, India) in accordance with the South African National Malaria Diagnosis Quality Assurance guidelines [42, 43]. The test was performed on individuals presenting with fever at the PHCs. The presence of P. falciparum parasites was confirmed by multiplex polymerase chain reaction (PCR) as previously described [44].
G6PD phenotypic screening
The CareStart™ G6PD RDT (Access Bio Inc., New Jersey, USA) was administered concurrently with the malaria RDT on 248 participants at the PHCs as per the manufacturers’ guidelines. Briefly, a finger prick blood sample (2 µl) was collected through the sample pipette, applied into the sample well and 2 drops (~ 100 µl) of assay buffer added. The test was developed at room temperature (25 °C) for 10 min, after which the results were read by two blinded primary evaluators, with a third blind evaluator acting as a tiebreaker to make the final decision in case of discrepancies. Normal G6PD individuals were classified according to the development of a distinct purple coloured test band, while in G6PD-deficient patients, no purple coloured test band developed. A test was declared invalid when either no or incomplete blood migration took place.
G6PD genotyping
DNA was extracted from blood-saturated filter paper discs of 3 mm diameter using the Chelex method [45]. Specific regions of the G6PD gene were amplified and subjected to restriction fragment length polymorphisms (RFLP, all restriction enzymes sourced from New England Biolabs, Inc., USA) to detect mutations as previously described [14]. A 308 base pair (bp) amplicon was amplified from genomic DNA and subjected to FokI digestion for 60 min at 37 °C. Uncut fragments were classified as G6PD variant B, while samples where two fragments of 184 and 124 bp were produced, were classified as G6PD A+ variants. Only samples positive for G6PD A+ mutations underwent further PCR amplification and RFLP analysis to detect additional mutations. Subsequent digestion was performed with N1aIII (G202A; 211, 81 and 130 bp), BspEI (G542T; 130, 80 and 50 bp), BstNI (G680T; 115, 98 and 29 bp) and NciI (T968C; 282, 162 and 120 bp) for 60 min at 37 °C (except for BstNI where digestion occurred at 60 °C). All G6PD deficiency restriction products were resolved on 3% agarose/TAE gel with ethidium bromide staining. Images were captured on UVDoc HD2 transilluminator (UVITEC, Cambridge, UK).
CYP2D6 genotyping
Genotyping of CYP2D6*4 was performed by PCR amplification of a 309 bp segment of the CYP2D6 gene followed by overnight digestion with BstNI at 60 °C as previously described [46], resulting in a 309 bp fragment for wt/wt CYP2D6*4; 309, 201 and 108 bp fragments for wt/mt CYP2D6*4 or 201 and 108 bp fragments for mt/mt CYP2D6*4. CYP2D6*17 genotyping was performed by PCR amplification of a segment of the cyp2d6 gene of interest and subsequent overnight digestion with BtsCI as previously described [46], resulting in 407, 328, 79, 67 and 55 bp fragments for wt/wt. For CYP2D6*17 wt/mt the resulting fragment sizes were 407, 328, 79, 67 and 55 bp; mt/mt CYP2D6*17 resulted in 328, 79, 67 and 55 bp fragments. For all the CYP2D6 variants, the PCR and restriction products were resolved on 1.5% agarose/TAE with ethidium bromide staining gel. Images were captured on UVDoc HD2 transilluminator.
Sanger sequencing
All G6PD A− positive PCR products were confirmed with Sanger nucleotide sequencing. After purification with the NucleoSpin® Gel and PCR clean-up kit (Macherey–Nagel, Duren, Germany), samples resuspended in HiDi™ formamide were sequenced using an ABI PRISM BigDye® terminator V3.1 cycle sequencing kit and processed on ABI PRISM 3130xl and ABI PRISM 3500xl genetic analyzers (Applied Biosystems™/Life Technologies, Carlsbad, USA). BLAST queries were conducted for preliminary identifications of the sequences and thereafter analysed with MAFFT v 7.182 and CLC sequence viewer software.
Data analysis
Data were analysed with SPSS version 20 (IBM Corp. NY, USA). Descriptive statistics were used to report the demographics of the participants, with phenotype and genotype being presented per PHC. Differences in proportions of individuals carrying the G6PD deficiency, CYP2D6*4 and CYP2D6*17 wildtype and mutant alleles were compared by Chi squared (χ2) tests. Odds ratios (ORs) were used to assess any association of G6PD deficiency, CYP2D6*4 and CYP2D6*17 occurrence based on gender. To test for conformity with the Hardy–Weinberg equilibrium (HWE), allele frequencies were analysed both by PHC and overall. The expected female allele frequencies were calculated based on male genotype prevalence (A+, A− and B) then compared with observed values using Chi squared tests as described [14]. The negative predictive value (NPV) of the CareStart™ G6PD deficiency kit was determined using the A− variant PCR–RFLP genotyping results as the ‘gold standard’ [47]. Agreement between the two test results was evaluated using Cohen’s Kappa coefficient (κ), with the following scale used to score strength of agreement; 0.01−0.20 slight; 0.21–0.40 fair; 0.41–0.60 moderate; 0.61–0.80 substantial and 0.81–1.00 almost perfect [48]. A P value of < 0.05 was considered statistically significant.
Results
Study participant characteristics and malaria parasite detection
From the six PHCs, a total of 248 participants were recruited into the study. More female participants (n = 193, 78%) compared to male participants (n = 55, 22%) were enrolled, with study participants’ median ages of 31 years for females (range 1–94) and 21 years for males (range 1–81). The majority (97%, 241/248) of participants self-identified as South African Venda ethnicity. Other ethnic groups screened included Pedi (1.2%, 3/248) and Tsonga (0.4%, 1/248), with 1.2% (3/248) Shona from Zimbabwe. None of the participants harboured detectable P. falciparum parasites by either P. falciparum RDT or single-step, multiplex PCR.
G6PD genotyping
The prevalence of the A+ and A− African G6PD variants among the study participants was determined by genotyping. Of the 248 participants screened, just over half (52%, 129/248) carried the G6PD normal B allele (Fig. 2a). The A376G mutation in exon V was detected in 32% (79/248) of the participants, identifying these individuals as carrying the A+ variant. A total of 13% (32/248) of the participants were G6PD A− heterozygous individuals. Only a minority of participants (3.2%, 8/248), were hemizygous or homozygous for the G6PD A− (A376G/G202A) variant. Among males, G6PD A− hemizygous individuals constituted 11% (6/55), while among females, 1% were G6PD A− homozygous (2/193). All G6PD A− variants identified contained A376G/G202A mutations, which was independently confirmed by DNA sequencing. The A542T, G680T or T968C mutations were not detected in the study population.

G6PD genotype prevalence. a G6PD genotypes (B patterned bar; A+ (A375G) white bar; A− (A375G/G202A) heterozygous light grey bar, hemizygous and homozygous dark grey bar) identified with PCR–RFLP and confirmed by sequencing for all the study participants. b G6PD deficiency stratified for each of the 6 primary health care clinics in Vhembe District, Limpopo Province, South Africa. (B patterned wedge; A+ (A375G) white wedge; A− (A375G/G202A) heterozygous light grey wedge, hemizygous and homozygous dark grey wedge)
When comparing the proportions of participants with G6PD A+ and A− variants in the population, considerable variations were observed among localities (P = 0.01 for A+ and P = 0.05 for A− genotypes, respectively). In Manenzhe PHC, the highest proportion (64%) of G6PD B variants but lowest proportion (8%) of G6PD A− variants were detected. All the A− participants from Madimbo and Manenzhe PHC were A− heterozygous females (Fig. 2b). Mulala PHC recorded the highest proportions of A− hemizygous participants (11%) while homozygous participants (6%) were only identified from Tshipise PHC. All of the A− deficient participants G6PDd were of Venda ethnicity.
The genotypic frequencies were tested to confirm adherence to the HWE. According to the principle, the genetic variation in a population should remain constant from generation to generation in the absence of disturbing factors. The HWE measurement indicated that in general, female gene frequencies did not significantly differ from predicted male gene frequencies (χ2 > 0.05). However, departures from HWE were seen for the G6PD deficiency variants for the general population (χ2 = 0.007) and in Tshipise PHC (χ2 = 0.009) (Table 1) where all of the study homozygous participants were reported. When comparing G6PDd A + and G6PDd A- genotypes, no distinctive association with gender was observed (P = 0.107 and 0.441, respectively).
Phenotypic G6PD screening
The CareStart™ G6PD deficiency RDT test kit was used for phenotypic evaluation of G6PD status in the test population, to explore its utility as point-of-care tool. The CareStart™ G6PD deficiency RDT identified 13% (33/248) of participants as G6PD-deficient (Table 2), with males showing a higher prevalence of G6PD deficiency (18%; 10/55) compared to females at 12% (23/193) (odds ratio 1.642; 95% confidence interval 0.7293–3.6992, P = 0.229). All the identified G6PD-deficient individuals bar one (Tsonga) were of Venda ethnic origin.
The CareStart™ G6PD deficiency RDT was able to detect all the genetically-determined G6PD-deficient hemizygous males and homozygous females (Table 2) with the highest risk of haemolysis. Comparison of the RDT phenotyping results with that of the G6PD A− (A376G/G202A) PCR–RFLP genotyping indicated specificity of 91.3% (95% CI 86.7–94.8) with a fair Cohen’s Kappa agreement (κ = 0.310). A more substantial agreement was observed among the males (κ = 0.711) than among the females (κ = 0.202). The negative predictive value for identifying hemizygous, homozygous and heterozygous participants by RDT was 0.88 (95% CI 0.85–0.91) compared to genotyping.
Cytochrome P450 genotype and phenotype distribution
Mutations in the cyp2d6 gene that could interfere with PQ activation were evaluated on the 248 collected samples (Table 3). Allele frequencies of 0.03 and 0.19 were detected for the CYP2D6*4 and CYP2D6*17 alleles, respectively. Only 2 of the study participants (0.8%, 2/248) were classified as mt/mt PM with activity scores (AS) of 0 with a minor proportion (6%, 15/248) classified as mt/mt IM (AS of 0.5–1.0), using the AS metric as described [49]. The majority of the population (231/248, 93%) was classified as extensive or normal metabolizers. No association between gender and either the CYP2D*4 (χ2, P = 0.731) or CYP2D6*17 (χ2, P = 0.906) genotype was noted.
Discussion
This study reports on the prevalence of G6PD deficiency as well as CYP2D6*4 and CYP2D7*17 mutations in a southern African malaria pre-elimination setting, with the aim of informing a low-dose PQ policy. The G6PD A− prevalence of 11% among hemizygous males was comparable to that previously reported in South African Venda ethnic group [50]. Higher G6PD deficiency prevalence among males was previously reported in ethnic groups residing in close proximity to neighbouring Mozambique and Zimbabwe [30, 50], consistent with the observations in this study. These countries have higher malaria transmission intensity compared to South Africa, which could explain their higher G6PD deficiency prevalence of up to 19% in males [51, 52]. Modelling the median population estimates for G6PD allele frequency predict low frequencies of 3.3% for South Africa [15]. However, maps aggregate average frequency data to national levels and may mask sub-national variation and local heterogeneity in G6PD deficiency. By contrast, in Swaziland, a study on PQ pharmacovigilance in two health facilities reported the absence of G6PD deficiency amongst 102 participants [53].
The presence of A− (A376G/G202A) variant was confirmed in G6PD-deficient individuals, corroborating its previously-described widespread distribution [29, 50]. Other G6PD variants such as the A542T, G680T or T968C mutations previously reported in the Gambia and Senegal [17, 18], were not identified in the current southern African study population. This is consistent with findings from two large randomized controlled multicentre studies in Africa [14, 54]. While the study aimed to inclusively genotype different G6PD deficiency variants identified as important in African settings, the presence of additional G6PD deficiency variants in the locality cannot be ruled out. Future studies using more novel and robust genotyping methods are envisaged, as they could identify novel mutations that may impact PQ roll-out [32].
PQ is an important addition to standard malaria treatment regimen to aid in transmission reduction, especially for countries on the cusp of elimination [6]. PQ efficacy may be influenced by individual and ethnic genetic differences amongst populations [21, 55] and is reliant on metabolic activation. The PM and IM CYP2D6 phenotypes observed (3.2 and 19.5%, respectively) may result in PQ treatment failure at the recommended 0.25 mg/kg dose [56]. These findings on CYP2D6*4 allele frequency and predicted PM phenotype is consistent with earlier reported 1.3% [31] and 4% [57] prevalence among the Venda ethnic group. Indigenous African blacks have consistently been reported with a high frequency of the CYP2D6*17 allele, in agreement with reported data in this study [31, 33, 58].
As South Africa moves towards malaria elimination, novel interventions such as the WHO-recommended low-dose PQ strategy need further exploration. However, individuals with the CYP2D6*4 variants that may be unresponsive to PQ treatment and CYP2D*17 variants, who may require higher PQ dose to ensure therapeutic efficacy, is cause for concern in the country [56]. While there is little conclusive evidence on the effect of PQ on community–wide transmission if deployed in mass drug administration [59], efficacy studies point to marked reduction in duration of gametocyte carriage [4, 60]. The PQ-associated gametocyte clearance and its outcomes on mosquito infectiousness depends on malaria transmission settings and the choice of artemisinin combination therapy. It has been demonstrated that PQ has modest benefits on asymptomatic individuals with lower parasite densities prior to initiation of combination PQ treatment with either artemether-lumefantrine [26] or dihydoartemisinin-piperaquine [60]. That PQ should not be administered to infants aged < 6 months, breastfeeding women of infants aged < 6 months as well as pregnant women (South Africa has a 12% teenage pregnancy rate and higher fertility rates among non-urban women) [61], identifies the treatment gaps for PQ, with impact dependent on the proportion of the population treated [62].
Accumulating evidence from Africa [63,64,65] and Asia [66], suggests that single-dose PQ at 0.25 mg/kg as a gametocytocidal drug to block transmission would be safe in areas where G6PD deficiency A− and other type 2 variants predominates, comparable to the settings in this study site. Encouraging evidence from neighbouring Swaziland, a country with a similar G6PD deficiency prevalence profile to South Africa, suggests the roll-out of a low-dose PQ policy with no prior genetic profiling requirement, at least in none G6PD-deficient participants by RDT, is associated with good safety profiles [53]. However, stringent pharmacovigilance and patient monitoring systems should be put in place to mitigate any adverse events. Further, the low levels of CYP2D6 PM and IM is encouraging for the use of single low-dose PQ as an adjunct to current malaria control tools, and may accelerate efforts to eliminate malaria in South Africa.
This study was limited by the small sample size constrained by the availability of the G6PD RDT tests. Moreover, a potential selection bias towards females (78%) was present in the sample set, and findings may, therefore, not be directly extrapolated to estimate the overall population structure and deficiency prevalence in the study area, which can be rectified through more general population sampling, rather than sampling only at PHC facilities. Since the study only deployed a G6PD deficiency RDT, enzymatic activity validation is ultimately required to evaluated functional loss of G6PD activity to disentangle female heterozygotes from deficient and the non-deficient females. Female gender segregation would control for the profound effects of comparing male hemizygous and female heterozygous on diagnostic performance of G6PD tests [67].
Conclusions
Phenotypic and genotypic analysis both detected a low prevalence of individuals carrying the G6PD A−, CYP2D6*4 PM variants and modest prevalence of the CYP2D6*17 IM variants in the study area. These preliminary findings suggest the deployment of single low-dose PQ poses a minimal risk to the area populations and could be useful in elimination strategies, in localized areas of South Africa.
References
World Health Organization. Eliminating malaria. Geneva: World Health Organization; 2016.
Moonasar D, Maharaj R, Kunene S, Candrinho B, Saute F, Ntshalintshali N, et al. Towards malaria elimination in the MOSASWA (Mozambique, South Africa and Swaziland) region. Malar J. 2016;15:419.
World Health Organization. Policy brief on single-dose primaquine as gametocytocide in Plasmodium falciparum malaria. Geneva: World Health Organization; 2015.
Dicko A, Brown JM, Diawara H, Baber I, Mahamar A, Soumare HM, et al. Primaquine to reduce transmission of Plasmodium falciparum malaria in Mali: a single-blind, dose-ranging, adaptive randomised phase 2 trial. Lancet Infect Dis. 2016;16:674–84.
Eziefula AC, Bousema T, Yeung S, Kamya M, Owaraganise A, Gabagaya G, et al. Single dose primaquine for clearance of Plasmodium falciparum gametocytes in children with uncomplicated malaria in Uganda: a randomised, controlled, double-blind, dose-ranging trial. Lancet Infect Dis. 2014;14:130–9.
Ashley EA, Recht J, White NJ. Primaquine: the risks and the benefits. Malar J. 2014;13:418.
Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008;371:64–74.
Minucci A, Moradkhani K, Hwang MJ, Zuppi C, Giardina B, Capoluongo E. Glucose-6-phosphate dehydrogenase (G6PD) mutations database: review of the “old” and update of the new mutations. Blood Cells Mol Dis. 2012;48:154–65.
Howes RE, Piel FB, Patil AP, Nyangiri OA, Gething PW, Dewi M, et al. G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: a geostatistical model-based map. PLoS Med. 2012;9:e1001339.
Ruwende C, Khoo S, Snow R, Yates S, Kwiatkowski D, Gupta S, et al. Natural selection of hemi-and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995;376:246–9.
Guindo A, Fairhurst RM, Doumbo OK, Wellems TE, Diallo DA. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med. 2007;4:e66.
Clarke GM, Rockett K, Kivinen K, Hubbart C, Jeffreys AE, Rowlands K, et al. Characterisation of the opposing effects of G6PD deficiency on cerebral malaria and severe malarial anaemia. Elife. 2017;6:e15085.
Mbanefo EC, Ahmed AM, Titouna A, Elmaraezy A, Trang NT, Phuoc Long N, et al. Association of glucose-6-phosphate dehydrogenase deficiency and malaria: a systematic review and meta-analysis. Sci Rep. 2017;7:45963.
Carter N, Pamba A, Duparc S, Waitumbi JN. Frequency of glucose-6-phosphate dehydrogenase deficiency in malaria patients from six African countries enrolled in two randomized anti-malarial clinical trials. Malar J. 2011;10:241.
Howes RE, Dewi M, Piel FB, Monteiro WM, Battle KE, Messina JP, et al. Spatial distribution of G6PD deficiency variants across malaria-endemic regions. Malar J. 2013;12:418.
Luzzatto L, Nannelli C, Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Hematol Oncol Clin North Am. 2016;30:373–93.
Clark TG, Fry AE, Auburn S, Campino S, Diakite M, Green A, et al. Allelic heterogeneity of G6PD deficiency in West Africa and severe malaria susceptibility. Eur J H Genet. 2009;17:1080–5.
De Araujo C, Migot-Nabias F, Guitard J, Pelleau S, Vulliamy T, Ducrocq R. The role of the G6PD AEth376G/968C allele in glucose-6-phosphate dehydrogenase deficiency in the seerer population of Senegal. Haematologica. 2006;91:262–3.
Pybus BS, Marcsisin SR, Jin X, Deye G, Sousa JC, Li Q, Caridha D, et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar J. 2013;12:212.
Johansson I, Ingelman-Sundberg M. Genetic polymorphism and toxicology–with emphasis on cytochrome p450. Toxicol Sci. 2011;120:1–13.
Marcsisin SR, Reichard G, Pybus BS. Primaquine pharmacology in the context of CYP 2D6 pharmacogenomics: current state of the art. Pharmacol Ther. 2016;161:1–10.
Human cytochrome P450 (CYP) allele nomenclature database. [https://www.pharmvar.org/htdocs/archive/cyp2d6.htm]. Accessed 24 Nov 2017.
Bains RK. African variation at Cytochrome P450 genes. Evolutionary aspects and the implications for the treatment of infectious diseases. Evol Med Public Health. 2013;2013:118–34.
Bennett JW, Pybus BS, Yadava A, Tosh D, Sousa JC, McCarthy WF, et al. Primaquine failure and cytochrome P-450 2D6 in Plasmodium vivax malaria. N Engl J Med. 2013;369:1381–2.
Masimirembwa C, Persson I, Bertilsson L, Hasler J, Ingelman-Sundberg M. A novel mutant variant of the CYP2D6 gene (CYP2D617) common in a black African population: association with diminished debrisoquine hydroxylase activity. Br J Clin Pharmacol. 1996;42:713–9.
Gonçalves BP, Pett H, Tiono AB, Murry D, Sirima S, Niemi M, et al. Age, weight, and CYP2D6 genotype are major determinants of primaquine pharmacokinetics in African children. Antimicrob Agents Chemother. 2017;61:e02590-16.
von Seidlein L, Auburn S, Espino F, Shanks D, Cheng Q, McCarthy J, et al. Review of key knowledge gaps in glucose-6-phosphate dehydrogenase deficiency detection with regard to the safe clinical deployment of 8-aminoquinoline treatment regimens: a workshop report. Malar J. 2013;12:112.
Dandara C, Swart M, Mpeta B, Wonkam A, Masimirembwa C. Cytochrome P450 pharmacogenetics in African populations: implications for public health. Expert Opin Drug Metab Toxicol. 2014;10:769–85.
Hitzeroth H, Bender K. Erythrocyte G-6-PD and 6-PGD genetic polymorphisms in South African Negroes, with a note on G-6-PD and the malaria hypothesis. Hum Genet. 1980;54:233–42.
Jenkins T. Genetic polymorphisms of public-health significance in southern Africa. S Afr J Sci. 1975;71:44–9.
Dandara C, Masimirembwa CM, Magimba A, Sayi J, Kaaya S, Sommers DK, et al. Genetic polymorphism of CYP2D6 and CYP2C19 in east-and southern African populations including psychiatric patients. Eur J Clin Pharmacol. 2001;57:11–7.
Dodgen TM, Hochfeld WE, Fickl H, Asfaha SM, Durandt C, Rheeder P, et al. Introduction of the AmpliChip CYP450 test to a South African cohort: a platform comparative prospective cohort study. BMC Med Genet. 2013;14:1.
Dodgen TM, Labuschagne CD, van Schalkwyk A, Steffens FE, Gaedigk A, Cromarty AD, et al. Pharmacogenetic comparison of CYP2D6 predictive and measured phenotypes in a South African cohort. Pharmacogenomics. 2016;16:566–72.
Statistics South Africa. Community Survey 2016, Statistical release P0301. Pretoria: Statistics South Africa; 2016.
Khosa E, Kuonza LR, Kruger P, Maimela E. Towards the elimination of malaria in South Africa: a review of surveillance data in Mutale Municipality, Limpopo Province, 2005 to 2010. Malar J. 2013;12:7.
Raman J, Morris N, Frean J, Brooke B, Blumberg L, Kruger P, et al. Reviewing South Africa’s malaria elimination strategy (2012–2018): progress, challenges and priorities. Mal J. 2016;15:438.
Blumberg L, Frean J. Malaria reduces globally but rebounds across southern Africa. S Afr J Infect Dis. 2017;32:3–4.
National Institute for Communicable Diseases. Communicable Diseases Communiqué 2017. Johannesburg: National Institute for Communicable Diseases; 2017.
Mpandeli S. Managing climate risks using seasonal climate forecast information in Vhembe District in Limpopo Province, South Africa. J Sust Dev. 2014;7:68.
Nenwiini S, Kabanda TA. Trends and variability assessment of rainfall in Vhembe South Africa. J Hum Ecol. 2013;42:171–6.
Massyn N, Day C, Dombo M, Barron P, English R, Padarath A. District health barometer 2012/13. Durban: Health Systems Trust; 2013. p. 1–420.
Blumberg LH. Recommendations for the treatment and prevention of malaria: update for the 2015 season in South Africa. S Afr Med J. 2015;105:175–8.
South African National Department of Health. National Malaria Diagnosis Quality Assurance Guidelines. Pretoria: South African National Department of Health; 2011.
Padley D, Moody A, Chiodini P, Saldanha J. Use of a rapid, single-round, multiplex PCR to detect malarial parasites and identify the species present. Ann Trop Med Parasitol. 2003;97:131–7.
Baidjoe A, Stone W, Ploemen I, Shagari S, Grignard L, Osoti V, et al. Combined DNA extraction and antibody elution from filter papers for the assessment of malaria transmission intensity in epidemiological studies. Malar J. 2013;12:272.
Naveen AT, Adithan C, Soya SS, Gerard N, Krishnamoorthy R. CYP2D6 genetic polymorphism in South Indian populations. Biol Pharm Bull. 2006;29:1655–8.
Kim S, Nguon C, Guillard B, Duong S, Chy S, Sum S, et al. Performance of the CareStart™ G6PD deficiency screening test, a point-of-care diagnostic for primaquine therapy screening. PLoS ONE. 2011;6:e28357.
Landis JR, Koch GG. The measurement of observer agreement for categorical data. Biometrics. 1977;33(1):159–74.
Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther. 2008;83:234–42.
Bernstein R. Occurrence and clinical implications of red-cell glucose-6-phosphate dehydrogenase deficiency in South African racial groups. S Afr Med J. 1963;37:447–51.
Reys L, Manso C, Stamatoyannopoulos G. Genetic studies on southeastern Bantu of Mozambique. I. Variants of glucose-6-phosphate dehydrogenase. Am J Hum Genet. 1970;22:203–15.
Galatas B, Mabote L, Simone W, Matambisso G, Nhamussua L, Manu-Pereira MD, et al. Heterogeneity of G6PD deficiency prevalence in Mozambique: a school-based cross-sectional survey in three different regions. Malar J. 2017;16:36.
Poirot E, Soble A, Ntshalintshali N, Mwandemele A, Mkhonta N, Malambe C, et al. Development of a pharmacovigilance safety monitoring tool for the rollout of single low-dose primaquine and artemether-lumefantrine to treat Plasmodium falciparum infections in Swaziland: a pilot study. Malar J. 2016;15:384.
Nguetse CN, Meyer CG, Adegnika AA, Agbenyega T, Ogutu BR, Kremsner PG, et al. Glucose-6-phosphate dehydrogenase deficiency and reduced haemoglobin levels in African children with severe malaria. Malar J. 2016;15:346.
Teh LK, Bertilsson L. Pharmacogenomics of CYP2D6: molecular genetics, interethnic differences and clinical importance. Drug Metab Pharmacokinet. 2012;27:55–67.
Potter BM, Xie LH, Vuong C, Zhang J, Zhang P, Duan D, et al. Differential CYP 2D6 metabolism alters primaquine pharmacokinetics. Antimicrob Agent Chemotherap. 2015;59:2380–7.
Sommers DK, Moncrieff J, Avenant J. Non-correlation between debrisoquine and metoprolol polymorphisms in the Venda. Hum Toxicol. 1989;8:365–8.
Wennerholm A, Dandara C, Sayi J, Svensson JO, Abdi YA, Ingelman-Sundberg M, et al. The African-specific CYP2D6* 17 allele encodes an enzyme with changed substrate specificity. Clin Pharmacol Ther. 2002;71:77–88.
Graves PM, Gelband H, Garner P. Primaquine or other 8-aminoquinoline for reducing Plasmodium falciparum transmission. Cochrane Database Syst Rev. 2015. https://doi.org/10.1002/14651858.CD008152.pub4.
Okebe J, Bousema T, Affara M, Di Tanna GL, Dabira E, Gaye A, et al. The gametocytocidal efficacy of different single doses of primaquine with dihydroartemisinin-piperaquine in asymptomatic parasite carriers in The Gambia: a randomized controlled trial. EBioMedicine. 2016;13:348–55.
South African National Department of Health, South African Medical Research Council, and ICF. South Africa Demographic and Health Survey 2016: key indicators. Pretoria: South African National Department of Health, South African Medical Research Council, and ICF; 2017.
Brady OJ, Slater HC, Pemberton-Ross P, Wenger E, Maude RJ, Ghani AC, et al. Role of mass drug administration in elimination of Plasmodium falciparum malaria: a consensus modelling study. Lancet Glob Health. 2017;7:e680–7.
Bastiaens GJH, Tiono AB, Okebe J, Pett HE, Coulibaly SA, Goncalves BP, et al. Safety of single low-dose primaquine in glucose-6-phosphate dehydrogenase deficient falciparum-infected African males: two open-label, randomized, safety trials. PLoS ONE. 2018;13:e0190272.
Mwaiswelo R, Ngasala BE, Jovel I, Gosling R, Premji Z, Poirot E, et al. Safety of a single low-dose of primaquine in addition to standard artemether-lumefantrine regimen for treatment of acute uncomplicated Plasmodium falciparum malaria in Tanzania. Malar J. 2016;15:316.
Tine RC, Sylla K, Faye BT, Poirot E, Fall FB, Sow D, et al. Safety and efficacy of adding a single low dose of primaquine to the treatment of adult patients with Plasmodium falciparum malaria in Senegal, to reduce gametocyte carriage: a randomized controlled trial. Clin Infect Dis. 2017;65:535–43.
Bancone G, Chowwiwat N, Somsakchaicharoen R, Poodpanya L, Moo PK, Gornsawun G, et al. Single low dose primaquine (0.25 mg/kg) does not cause clinically significant haemolysis in G6PD deficient subjects. PLoS ONE. 2016;11:e0151898.
Johnson MK, Clark TD, Njama-Meya D, Rosenthal PJ, Parikh S. Impact of the method of G6PD deficiency assessment on genetic association studies of malaria susceptibility. PLoS ONE. 2009;4:e7246.
Authors’ contributions
SSA, JR and LMB conceived and designed the study, PK, JF and JR coordinated field sample and patient information collection. SSA and TM performed the genotyping and phenotyping assays. SSA, JR, TB, JN and LMB wrote and revised the paper. All authors read and approved the final manuscript.
Acknowledgements
We thank staff of the Malaria Control Programme, Vhembe District, Limpopo Province, who assisted with field data collection as well as the communities of Mutale and Musina local municipalities and primary health care facilities staff for their support.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analysed during the current study are available upon reasonable request from the corresponding author.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Ethical approval for the study was obtained from the ethical review committees of the University of Pretoria (Ethics reference No. 406/2014) and the Limpopo Department of Health (Ref 4/2/2). Study participants’ or their guardians gave informed consent before being enrolled in the study.
Funding
The South African Medical Research Council (SA MRC) is acknowledged for a Self-Initiated Research grant (to JN) and funding of the UP ISMC and WRIM as Collaborating Centres for Malaria Research. The South African Research Chairs Initiative of the Department of Science and Technology, administered through the South African National Research Foundation is gratefully acknowledged for support to LMB (UID84627). JN was supported through an International Society for Infectious Diseases grant.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Awandu, S.S., Raman, J., Makhanthisa, T.I. et al. Understanding human genetic factors influencing primaquine safety and efficacy to guide primaquine roll-out in a pre-elimination setting in southern Africa. Malar J 17, 120 (2018). https://doi.org/10.1186/s12936-018-2271-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-018-2271-z