
Abstract
Background
The emergence of multidrug-resistant strains of Plasmodium falciparum poses a great threat of increased fatalities in cases of cerebral and other forms of severe malaria infections in which parenteral artesunate monotherapy is the current drug of choice. The study aimed to investigate in a mouse model of human cerebral malaria whether a trioxaquine chemically synthesized by covalent linking of a 4,7-dichloroquinoline pharmacophore to artesunate through a recent drug development approach termed ‘covalent bitherapy’ could improve the curative outcomes in cerebral malaria infections.
Methods
Human cerebral malaria rodent model, the C57BL/6 male mice were infected intraperitoneally (ip) with Plasmodium berghei ANKA and intravenously (iv) treated with the trioxaquine from day 8 post-infection (pi) at 12.5 and 25 mg/kg, respectively, twice a day for 3 days. Treatments with the trioxaquine precursors (artesunate and 4,7-dichloroquine), and quinine were also included as controls. In vivo safety evaluation for the trioxaquine was done according to Organization for Economic Co-operation and Development (OECD) guidelines 423, where female Swiss albino mice were orally administered with either 300 or 2000 mg/kg of the trioxaquine and monitored for signs of severity, and or mortality for 14 days post-treatment.
Results
The trioxaquine showed a potent and a rapid antiplasmodial activity with 80% parasite clearance in the first 24 h for the two dosages used. Long-term parasitaemia monitoring showed a total parasite clearance as the treated mice survived beyond 60 days post-treatment, with no recrudescence observed. Artesunate treated mice showed recrudescence 8 days post-treatment, with all mice in this group succumbing to the infection. Also, 4,7-dichloroquinoline and quinine did not show any significant parasitaemia suppression in the first 24 h post-treatment, with the animals succumbing to the infection.
Conclusion
Covalent bitherapy proves to be a viable source of urgently needed new anti-malarials for management of cerebral malaria, and this polypharmacology approach could be a potential strategy to protect artesunate from parasite resistance and in potentially improving clinical outcomes in severe forms of malaria infections.
Similar content being viewed by others


Background
Despite many years of research and great progress in the fight against malaria [1], cerebral malaria (CM) has remained the most dreadful severe complication of Plasmodium falciparum, with a higher burden greatly felt in sub-Saharan Africa [2], a continent that already battles the effect of other diseases such as tuberculosis and HIV/AIDS [3]. Children 5 years old and younger are the most susceptible, with a staggering 90% CM related fatalities [4]. With no effective vaccine against malaria yet, chemotherapy has remained the mainstay of malaria management [5]. Extremely limited options for chemotherapy in CM have been a great challenge [5]. For decades, quinine has been the drug of choice for management of CM [6]. Concerns over increasing resistance to the quinoline-based drugs such as chloroquine (CQ) prompted World Health Organization (WHO) policy change to use of artemisinin-based drugs, such as artemether and artesunate. In management of CM, preference has been given to parenteral artesunate due to its superior efficacy and survival outcome in comparison to intramuscular artemether [7, 8]. Despite effective CM chemotherapy with artesunate, mortality rate still remains as high as 15–25% among treated children while those that survive the acute episodes of CM suffer long-term neurological sequelae including epilepsy [9].
Plasmodium falciparum, the most lethal pathogen among the Plasmodium species causing up to 90% of all malaria-related deaths in sub-Saharan Africa has shown multi-drug resistance [10]. Reports of parasite resistance against the WHO-recommended artemisinin (ART) derivatives—the first-line anti-malarials—have already been documented, prompting an urgent need for new drugs [10]. Extremely low success rates for new chemotherapy in the development pipeline to proceed to clinical trials [11] has necessitated a rational deployment of the available effective drugs to optimize their useful therapeutic life and also to protect them from being rendered ineffective due to parasite resistance [12]. Currently, ART derivatives are used in combination with longer durational anti-malarials in the artemisinin-based combination therapy (ACT) strategy, in order to protect them against resistance [5]. Unfortunately, in CM, artesunate has often been used as a monotherapy [7], thus calling for a polypharmacology strategy in such a scenario, especially in sub-Saharan Africa.
In the long and difficult search to develop effective, cheaper and urgently needed anti-malarial drugs, many research groups have employed the emerging strategy in medicinal chemistry called hybridization or covalent bitherapy [12,13,14]. This is a rational drug design approach based on covalent linking of drug pharmacophores into a single hybrid molecule, termed as hybrid drug, dual drug or conjugates [12]. The resulting hybrid drugs have been reported to possess superior efficacy to either the individual precursors alone or their co-formulations [15]. This approach reduces the risk of treatment failure and also offers resistance protection to the partner drug or to both drugs [16], and is believed to be cheaper since only the active moieties are covalently linked, thus avoiding the long, laborious and expensive drug development procedures of individual drugs before co-formulation [16]. Trioxaquines, which are hybrid drugs arising from conjugation of 1,2,4-endoperoxide bridge of the artemisinins and a quinoline pharmacophore, are demonstrative of the validity of this approach [12].
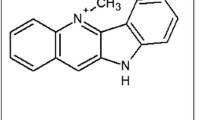
In this study, a trioxaquine termed N-(7-chloroquinolin-4-ylamino)-ethyl-artesunate-19-carboxamide (Fig. 1) was evaluated for curative effect of CM in mouse model. The dual drug was previously synthesized and demonstrated to possess remarkable antiplasmodial effect against blood stage rodent malaria parasite (ED50 and ED90 of 5.5 and 13.5 mg/kg, respectively) [17]. The dual drug had also shown a remarkable in vitro antiplasmodial activity against CQ-sensitive (D6) (IC50, 6.89 ng/mL) and CQ-resistant (W2) (IC50, 3.62 ng/mL) P. falciparum isolates [17].

Structure of trioxaquine: N-(7-chloroquinolin-4-ylamino)-ethyl-artesunate-19-carboxamide
Methods
Parasites, hosts and drugs
Plasmodium berghei (ANKA strain) was used in the study. This parasite, maintained at −80 °C in Kenya Medical Research Institute (KEMRI) at the Centre for Biotechnology, Research, and Development (CBRD), was thawed and revived by intraperitoneal (ip) inoculation into male Swiss albino mice. The mice served as parasite donor for passaging into C57BL/6 male mice weighing 25 ± 2 g, used as a mouse model of CM. The mice were subsequently randomized into groups of five per cage and fed with pellets and water, ad libitum [18].
The test drugs, namely artesunate, 4,7-dichloroquinoline and quinine were purchased from Sigma Aldrich®, while the trioxaquine, a kind gift from Prof. Francis W Muregi, was previously synthesized by linking the 4,7-dichloroquinoline to a linker and coupling the product to artesunate [17]. On the day of administration, artesunate, the dichloroquinoline and the trioxaquine were dissolved in a solution of 70% Tween-80 (d = 1.08 g/ml) and 30% ethanol (d = 0.81 g/ml) at a final diluted concentration of 7% (v/v), and 3% (v/v) for Tween 80 and ethanol, respectively [18]. Quinine was dissolved in normal saline.
Infection of experimental mice
The donor mice were anaesthetized and the infected red blood cells collected using heparinized syringe via cardiac puncture. The parasitaemia was then adjusted downwards using sterilized phosphate-buffered saline (PBS). Male C57BL/6 mice were then inoculated ip with approximately 1 × 105 parasitized erythrocytes (pRBCs) at inoculum of 0.2 ml. The infected mice were then randomized in plastic cages in groups of five and observed daily for signs of experimental cerebral malaria (ECM), including rough coat, limb paralysis, ataxia, convulsions, and or coma [19, 20]. In C57BL/6, ECM signs usually occur between day 6 and 9 post-infection (pi) [19, 20]. Giemsa-stained thin blood smear slides were also observed under light microscope (×1000) to establish infection.
In vivo evaluation of the trioxaquine in cerebral malaria rodent models
The treatment of CM in experimental mice with established infection was carried out by the approach of Ryley and Peters [21] in which treatment with a drug was initiated when the infection was already well established and the mice had exhibited clear signs for cerebral malaria. Based on the previous results of the 4-day suppressive test that had been used to evaluate chemosuppression ability of the trioxaquine in vivo [17], a dosage of 12.5 and 25 mg/kg were administered intravenously (iv) twice daily for 3 days through the lateral tail veins from day 8 pi. Three positive control groups were also similarly treated with artesunate (12.5 mg/kg), quinine (60 mg/kg), and 4,7-dichloroquinoline (12.5 mg/kg). A negative control group that received only the drug vehicle was also included. Parasitaemia of each mouse was determined by microscopic examination of Giemsa-stained, thin blood smears prepared from mouse tail blood. Slides were coded and infected red blood cell (iRBC) count was done microscopically in at least five microscopic fields, each with an approximation of 200–400 iRBCs. Smears were done at 24 h post-treatment, and 48 h post-treatment later to monitor progression of parasitaemia. Parasitaemia counts of blood films from each mouse were processed using Microsoft® Excel (Microsoft Corp.). Percentage (%) parasite suppression (chemosuppression) at each drug dose was determined as described by Muregi et al. [22]:
The survival of mice was monitored for up to 60 days after the end of the 3-day treatment. All RBC counts and parasitaemia levels were expressed as mean values ± standard deviations and the parasitaemia data were processed using the one-way analysis of variance (ANOVA). Mean parasitaemia between each experimental group relative to controls was compared using Student’s t test with a probability of 5% (p < 0.05) considered significant.
Evaluation of the integrity of mouse blood–brain barrier
C57BL/6 mice were injected iv with 200 μl of 2% (w/v) Evans Blue (Fischer Scientific®) dye on day 8 pi after the experimental animals had exhibited clear signs for cerebral malaria. The animals were euthanized after 1 h for brain extraction to assess the extent of brain staining with the dye and the images documented. The principle behind this method is based upon the formation of bonds between the dye and plasmatic albumin and, in the event of brain blood vessels leakage due to compromised blood–brain barrier (BBB), the dye–protein complex migrates to the tissue, impregnating it in a tone of blue visible to the naked eye [23].
Safety evaluation of the trioxaquine
To evaluate the safety of the trioxaquine, the OECD Test Guidelines 423 (Acute Toxic Class Method, adopted on 17 December 2001) were adopted. This is a protocol that makes use of three uninfected animals of same gender, especially females, at every dosage category and assessment based on death and or signs of severity observed with the dosed animals. Averagely, two to four dosage steps in the range of 5, 50, 300 and 2000 mg/kg are considered enough in making judgement on the acute oral toxicity of the test compound [24].
Two fixed dose levels of 300 and 2000 mg/kg were used and dosing was done in a stepwise manner starting with the lowest dose upwards. Swiss albino female mice weighing 20 ± 2 g were used where the animals were first fasted for 4 h, after which they were dosed orally with the above dosages. The start, duration and severity of toxic signs of the test compound, such as changes in skin and fur, eyes, mucous membranes, tremors, convulsions, salivation, diarrhoea, coma, and even death were monitored. The animals were observed individually at least once during the first 30 min, periodically during the first 24 h with special attention given during the first 4 h, and daily thereafter for the next 14 days.
Ethical statement
The use of mice in this research work was approved by KEMRI's Animal Care and Use Committee (ACUC) (Approval No: KEMRI/ACUC/01.04.16) and the Scientific Ethical Review Unit (SERU) (Permit No: KEMRI/RES/7/3/1/2016). All mice that were deemed to have completed their intended role in the study were euthanized using 150 mg/kg body weight sodium pentobarbital solution injected ip, placed in biohazard bags and incinerated.
Data analysis
Data analysis was performed using SPSS version 16. All values are expressed in means (±SDs). Cumulative survival rates were calculated according to Kaplan–Meier method and groups were compared using the log-rank test. Parasitaemia courses were compared by analysis of variance (ANOVA). Parasitaemia before and at 24 h after treatment were compared by paired Student t test. P value <0.05 was considered to be statistically significant.
Results
Assessment of the therapeutic effect of the trioxaquine
Treatment was initiated on day 8 pi after the experimental animals were clearly diagnosed with ECM or having presented with at least one or both well-recognized signs of ECM in animal models, including neurological perturbations such as ataxia, rough coat, limb paralysis, inter alia. Parasitaemia load reduction for each test drug within the first 24 h post-treatment is presented in Table 1. The parasitaemia was monitored for 60 days post-treatment.
At the two dosages used, 12.5 and 25 mg/kg iv, the trioxaquine showed a strong and promising antiplasmodial activity. At 12.5 mg/kg, parasitaemia decreased from 11.7 to 1.6 (86.6% clearance) and from 11.7 to 0.4 (96.4% clearance) at 25 mg/kg just after 24 h post-treatment and no parasite was observable under microscope at 48 h post-treatment. A potent anti-malarial activity was therefore exhibited by the artesunate–quinoline hybrid drug at the two dosages, resulting in a rapid parasite clearance. The treated animals also survived beyond 60 days post-treatment except for the 12.5 mg/kg, where death of one animal was recorded on day 4 post-treatment even after no parasite was observed under microscope.
Artesunate monotherapy at 12.5 mg/kg also showed a rapid parasitaemia clearance of 84.8% within the first 24 h post-treatment, and no parasite was observable under microscope at 48 h post-treatment. However, recrudescence was observed on day 8 post-treatment with all the animals succumbing to the infection by day 16 post-treatment (Fig. 3). Quinine at 60 mg/kg and 4,7-dichloroquinoline at 12.5 mg/kg did not show any significant parasitaemia reduction in the first 24 h post-treatment with all the animals in these experimental groups dying by day 12 and day 7 post-treatment, respectively (Fig. 3). Quinine, even after exhibiting a significant parasitaemia reduction in the 48th h post-treatment (44.9%), on further monitoring of parasitaemia showed progression of parasite growth for this group leading to death of mice (Fig. 3). Mice in untreated control group also succumbed to the infection by day 10 pi (Fig. 3).
Lack of recrudescence observed with the trioxaquine-treated animals could be attributed to the 4,7-dichloroquinoline, the partner drug to artesunate in the trioxaquine.
Assessment of integrity of mouse blood–brain barrier
Damage to the blood–brain barrier (BBB) has been implicated and is believed to be one of the underlying mechanisms of pathophysiology of CM, as the leaky BBB would allow toxic or unwanted compounds to enter the brain, causing neurological dysfunction. Observations of the brain tissue in infected animals that were never injected with the 2% Evans Blue dye on day 8 pi showed a higher degree of brain whitening in C57BL/6 mice (Fig. 2a). The massive brain whitening observed in Fig. 2a is believed to be due to reduced or blocked blood flow at multiple sites in the brain (hypoxia), a confirmation of the phenomenon of pRBC sequestration in the brain microvasculature, which is one of the major hallmarks of cerebral malaria. Uninfected C57BL/6 mice (Fig. 2b) showed a normal healthy brain with normal blood supply, an absence of brain staining with Evans Blue dye indicating an intact BBB integrity. Observation of brains after the injection of the infected experimental animals with 2% Evans Blue dye on day 8 pi showed a higher extent of brain staining in the mice (Fig. 2c), a feature that substantiates the impairment of the integrity of the BBB. Observation of brain images of infected C57BL/6 mice at 60 days post-treatment exhibited complete recovery, return of normal blood supply and lack of brain staining by the Evans Blue dye (Fig. 2d), results that are comparable to Fig. 2b of a healthy uninfected animal.

Evaluation of blood–brain barrier integrity with Evans Blue dye. The susceptible mice were first infected with P. berghei ANKA and the dye administered iv on day 8 pi after the infected mice had exhibited clear signs of ECM. The mice were euthanized 1 h later and the brain images documented. The same protocol was repeated on mice that were treated with trioxaquine at 60 days post-treatment. a Non-stained, P. berghei ANKA-infected C57BL/6 mouse brain on day 8 pi depicting hypoxia; b non-infected C57BL/6 mouse brain depicting normal blood supply; c brain staining for C57BL/6 mouse on day 8 pi; d depicts brain from the same animal model on day 60 post-treatment exhibiting recovery from experimental cerebral malaria, and lack of staining with Evans Blue dye
Evaluation of safety of trioxaquine in mice
Acute oral toxicity of trioxaquine was evaluated by using uninfected female mice that had been starved for 4 h, administered with oral dosages of 300 and 2000 mg/kg once. The test animals were then observed for 14 days post-dosing, basing the assessment on signs of severity, and/or death observed with test animals. All the animals did not show any sign of severity or toxicity as no changes in skin and fur, eyes and mucous membranes, as well as aberant respiratory activities were observed; also no tremors, convulsions, salivation, diarrhoea, sleep, and coma were observed. The mice survived past the 14 days of observation with 67% survival recorded with the dose of 2000 mg/kg. This is an indication that the exact lethal dose-50 (LD50) could be within 2000 mg/kg resulting to a possible therapeutic index (TI) value of >400, considering the oral lethal dose-50 to that of effective dose-50 (ED50) (i.e. LD50/ED50) ratio. The ED50 had previously been determined orally during in vivo antiplasmodial evaluation for the trioxaquine using the 4-day suppressive test [17]. This relatively weak toxicity indicates a very wide margin of safety for the test compound, implying that the compound could be a promising drug candidate for further exploration.
Discussion
Infection with P. falciparum often has high chance of progressing into CM, a severe form of malaria that contributes to high malaria mortality in sub-Saharan Africa [9]. Children 5 years old and younger, expectant women at first trimester, and individuals with naive immunity visiting malaria-endemic areas are at increased risk of infection [5].
Plasmodium falciparum has shown a rising trend of evolution and emergence of resistance to the currently used drugs, even to the WHO gold standard anti-malarial, ACT [5, 10]. Also, very few anti-malarial drugs are in the clinical development pipeline. Optimizing use of existing drugs as well as use of rational drug development strategies, such as covalent bitherapy, could lead to enhanced, useful therapeutic lives of existing therapy as well as add new therapy to the existing anti-malarial drug repertoire [12, 13]. In covalent bitherapy, just as in combination therapy, the goal is to either delay or circumvent development of resistance. In trioxaquines, the fast-acting precursor (ART pharmacophore) provides rapid clearance of the bulk of parasite load while the quinoline moiety clears the remnant parasite that survives the effect of the former, until complete clearance is achieved [13]. This strategy has potential to improve the therapeutic effectiveness as well as delaying or circumventing the emergence of resistance to both individual precursors of the hybrid drug, besides overcoming the challenge of a long drug development pipeline for co-formulated ACT drugs [15]. The trioxaquine exhibited a higher efficacy compared to individual precursors alone (Table 1). Based on the curative test using established infection, the trioxaquine though exhibiting activities comparable to artesunate, proved to be more effective than both the parenteral artesunate, quinine and 4,7-dichloroquine (Table 1). The trioxaquine manifested a rapid parasite clearance of greater than 80% within 24 h post-treatment and no parasite was observable under microscope at the 48 h post-treatment for both dosages of 12.5 and 25 mg/kg (Table 1).
Long-term monitoring of animals treated with trioxaquine at both 12.5 and 25 mg/kg showed no recrudescence, with the animals surviving beyond 60 days post-treatment (Fig. 3). This is a clear indication of complete cure in the treated animals since no recrudescence was observed, an indicator that the dual drug could potentially circumvent or delay development of drug resistance.

Survival probability analysis of the treated mice after infection with Plasmodium berghei ANKA, and treated on day 8 post-infection (pi) with various dosages of the trioxaquine, artesunate, 4,7-dichloroquine, and quinine. The mice were monitored for 60 days post-treatment. The experimental animals were first infected with an inoculum of 0.2 ml of approximately 1 × 105 parasitized erythrocytes with P. berghei ANKA, treated with 25 and 12.5 mg/kg of the test drug on day 8 pi, twice a day for 3 days and their survival monitored for 60 days post-treatment. Quinine at 60 mg/kg, artesunate at 12.5 mg/kg, 4,7-dichloroquinoline at 12.5 mg/kg, and untreated control groups were included. Survival probability analysis using Kaplan–Meier software revealed that there was a significant reduction in mortality in trioxaquine-treated groups (p < 0.05) in comparison to the standards (quinine, artesunate, 4,7-dichloroquinoline). Apart from the trioxaquine treatment groups, all the other treatment groups, as well as the negative controls, had 100% mortality by day 16 pi
Although rapid parasite suppression (84.8%) was observed in the first 24 h post-treatment with artesunate and no parasite was observable under microscope 48 h post-treatment, recrudescence was observed in this treatment group with all the animals succumbing to the infection by day 16 post-treatment. Even though the quinoline moiety, on the other hand, did not show any significant parasite reduction in the first 24 h post-treatment, its contribution in trioxaquine was manifested by lack of recrudescence in trioxaquine-treated animals even beyond 60 days post-treatment. The quinoline pharmachophore (4,7-dichloroquinoline) is believed to promote dual drug accumulation in the food vacuole, allowing the artemisinin-based partner (artesunate) to have a longer half-life [25], a possible explanation for the observed improved treatment outcome in the trioxaquine treatment groups compared to the artesunate-treated group. During treatment with 12.5 mg/kg of the trioxaquine, one mouse was lost on day 4 post-treatment. However, it was deemed that the death was not due to drug toxicity since even mice that received 25 mg/kg of trioxaquine had 100% survival. CM leads to a multi-organ dysfunction involving the liver, kidney and brain with the extent of the organs’ damage varying from individual to individual. Damage to these major organs are cited as the major reason behind CM deaths in infected individuals even after chemotherapeutic intervention [26]. This reason could support the loss of one animal in the 12.5-mg/kg trioxaquine treatment group even after parasitaemia monitoring had shown complete clearance. These results are in agreement with other previously related studies using the same experimental animals, which noted manifestation of neurological symptoms of CM between days 6 and 9 pi [18, 19, 27] and death in untreated animals by day 10 pi [28]. Recrudescence was also reported in artesunate-treated animals even at higher doses of 32 and 64 mg/kg, with all the treated animals succumbing to the infection and quinine being effective only at higher doses >120 mg/kg, which were not well tolerated by the experimental animals [29].
However, albeit in blood-stage infection, it was earlier reported that trioxaquines were capable of inhibiting parasites in mice and also affording curative effect in both the 4-day suppressive test and in established infection studies [13].
Two components have been implicated to be participating in the development of cerebral malaria, namely, the parasite-related factors and the host immune factors. Rupture of the hepatocytes leads to accumulation of the iRBCs within the brain microvasculature as the parasite invades the blood stream [30]. On the blood side of the BBB, P. falciparum-infected RBCs (PfRBCs) cytoadhere to the brain endothelium by binding of P. falciparum erythrocyte membrane protein 1 (PfEMP1), a specific cell-surface ligand expressed by iRBCs [31]. The sequestration of iRBCs triggers activation of the endothelial cells (ECs), which in turn lead to inflammatory responses where there is release of pro-inflammatory cytokine tumour necrosis factor (TNF) [32]. This cytokine is known to upregulate endothelial receptor cells, resulting in upregulation of intracellular adhesion molecules 1 (ICAM-1) and vascular cellular adhesion molecules-1 (VCAM–1) [33]. All these factors would eventually lead to numerous downstream vascular effects, such as increased vasoconstriction, reduced cerebral blood flow to several sites in the brain, vascular obstruction, oxygen starvation, and even disruption of the integrity of the BBB [34]. The observed whitening of the susceptible strain brains (Fig. 2a) could therefore be attributed to the reduced or the complete blockage of cerebral blood flow to several sites in the brain, a condition termed as hypoxia.
The BBB is comprised of specialized endothelial cells (ECs) that function as a selective permeable membrane to control nutrients and ion transport into the brain and to bar unwanted molecules or compounds into the brain [35, 36]. The ECs are sealed together by the presence of tight junctions. However, loss of tight junctions has been observed in areas of iRBC sequestration, leading to creation of openings or gaps within the BBB [36]. Such gaps or openings could function as a gateway for the entry of toxic molecules or compounds into the brain, thus a pointer to the dysfunctioning BBB, which is a major pathological event associated with CM [34, 37]. This was well demonstrated in this work through staining of the human cerebral malaria (HCM) mouse models’ brains with the 2% Evans Blue dye (Fig. 2c), an indicator of the impaired BBB integrity in the infected mice. The data of this study are similar to other previously related studies that confirmed impairment of the integrity of BBB in susceptible animal models and subsequent brain staining with Evans Blue dye [27, 38,39,40].
Good safety profile was exhibited as the drug was well tolerated in mice even at high dose of 2000 mg/kg administered in an acute oral toxicity test, with 67% survival. The remarkable mice survival indicates that the exact LD50 could be within 2000 mg/kg. However, it should be noted that this is a dosage 40 times the curative dose of 25 mg/kg. Considering the possible LD50/ED50 ratio, a possible TI value >400 reflects a wider margin of safety for the trioxaquine. The same safety profile was manifested in in vitro cytotoxicity studies where the selectivity index (SI) of the trioxaquine was determined by comparing the IC50s of Hep2 cell line and that of the CQ-resistant parasite (W2) for the trioxaquine [17]. The high SI value obtained (>2762) indicates that the high antiplasmodial activity for the trioxaquine observed during in vitro antiplasmodial evaluation was due to its activity and not due to its cytotoxicity [17]. A similar class of trioxaquine was reported to have antiplasmodial activity against all the erythrocytic forms of the parasite, including the gametocytes with TI value range of 23–100 considered safe [13]. The high safety profile exhibited both in vivo and in vitro by the trioxaquine would therefore be attributed to the fact that the individual precursors for the trioxaquine are already indicated for clinical use in the management of malaria.
The remarkable efficacy and good safety profile of the trioxaquine observed imply that if the findings could be replicated in clinical studies, then the drug has potential for use in management of CM.
Conclusion
The remarkable in vivo results for the trioxaquine obtained by the established infection test, as well as the post-treatment survival data, clearly indicate that covalent bitherapy is a viable strategy for development of future anti-malarial agents for management of CM. An encouraging safety profile of the drug in mice with a therapeutic index (TI) value of >400 is not surprising since the precursors are part of drugs already indicated for clinical use. Overall therefore, the curative effect together with good safety profiles observed with the trioxaquine clearly demonstrate its potential as a drug candidate for management of CM, especially in a time of shrinking anti-malarial armamentarium for management of CM.
Abbreviations
- PfiRBCS:
-
Plasmodium falciparum infected red blood cells
- NACOSTI:
-
National Commission for Science Technology and Innovation
- BBB:
-
Blood brain barrier
- CBRD:
-
Centre for Biotechnology Research and Development
- CTMDR:
-
Centre for Traditional Medicine and Drug Research
- ECs:
-
Endothelial cells
- ED50 :
-
Effective dose causing clearance of 50% of the parasite
- ICAM-1:
-
Intracellular adhesion molecule 1
- iRBCs:
-
Infected red blood cells
- CM:
-
Cerebral malaria
- LD50 :
-
Lethal dose causing death of 50% of test animals
- TI:
-
Therapeutic index
- TNF:
-
Tumour necrosis factor
- SI:
-
Selective index
- SERU:
-
Scientific Ethics Review Unit
- WHO:
-
World Health Organization
- VCAM-1:
-
Vascular cellular adhesion molecule 1
References
Wells TNC, Van Huijsduijnen RH, Van Voorhis WC. Malaria medicines, a glass half full. Nat Rev Drug Discov. 2015;14:424–5.
Desruisseaux MS, Machado FS, Weiss LM, Tanowitz HB, Golightly LM. Cerebral malaria: a vasculopathy. Am J Pathol. 2010;176:1075–8.
Mock NB, Duale S, Brown LF, Elliott S, et al. Conflict and HIV, a framework for risk assessment to prevent HIV in conflict-affected settings in Africa. Emerg Themes Epidemiol. 2004;1:6.
Boivin MJ, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, et al. Cognitive impairment after cerebral malaria in children: a prospective study. Pediatrics. 2007;119:360–6.
Schlitzer M. Antimalarial drugs—what is in use and what is in the pipeline. Arch Pharm (Weinheim). 2008;341:149–63.
Achan J, Talisuna AO, Erhart A, Yeka A, Tibenderana JK, Baliraine FN, et al. Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria. Malar J. 2011;10:144.
Pasvol G. Management of severe malaria: interventions and controversies. Infect Dis Clin North Am. 2005;19:211–40.
Phu NH, Tuan PQ, Day N, Mai NT, Chau TT, Chuong LV, et al. Randomized controlled trial of artesunate or artemether in Vietnamese adults with severe falciparum malaria. Malar J. 2010;9:97.
Idro R, Marsh K, John CC, Newton CR. Mechanisms of brain injury and strategies for improved neuro-cognitive outcome. Pediatr Res. 2010;68:267–74.
Phyo AP, Nkhoma S, Stepniewska K, Ashley EA, Nair S, McGready R, et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379:1960–6.
Olliaro P, Wells TN. The global portfolio of new antimalarial medicines under development. Clin Pharmacol Ther. 2009;85:584–95.
Muregi FW, Ishih A. Next-generation antimalarial drugs: hybrid molecules as a new strategy in drug design. Drug Dev Res. 2010;71:20–32.
Benoit-Vical F, Lelièvre J, Berry A, Deymier C, Dechy-Cabaret O, Cazelles J, et al. Trioxaquines are new antimalarial agents active on all erythrocytic forms, including gametocytes. Antimicrob Agents Chemother. 2007;51:1463–72.
Morphy R, Rankovic Z. Designed multiple ligands: an emerging drug discovery paradigm. J Med Chem. 2005;48:6523.
Grellepois F, Grellier P, Bonnet-Delpon D, Bégué J-P. Design, synthesis and antimalarial activity of trifluoromethylartemisinin-mefloquine dual molecules. Chem Biochem. 2005;6:648–52.
Coslédan F, Fraisse L, Pellet A, Guillou F, Mordmüller B, Kremsner P, et al. Selection of a trioxaquine as an antimalarial drug candidate. Proc Natl Acad Sci USA. 2008;105:17579–84.
Wamakima HN. Synthesis and antimalarial evaluation of a quinoline–trioxane hybrid drug. Kenyatta University. Retrieved from Kenyatta University e-repository. 2015. http://etd-library.ku.ac.ke/handle/123456789/13316. Accessed 8 Apr 2017.
Gimode WR, Kiboi DM, Kimani FT, Wamakima HN, Burugu MW, Muregi FW. Fitness cost of resistance for lumefantrine and piperaquine-resistant Plasmodium berghei in a mouse model. Malar J. 2015;14:38.
Wang M, Feng Y, Pang W, Qi Z, Zhang Y, Guo YJ, et al. Parasite densities modulate susceptibility of mice to cerebral malaria during co-infection with Schistosoma japonicum and Plasmodium berghei. Malar J. 2014;13:116.
Ghosh S, Sengupta A, Sharma S, Sonawat HM. Multivariate modelling with 1H NMR of pleural effusion in murine cerebral malaria. Malar J. 2011;10:330.
Ryley JF, Peters W. The antimalarial activity of some quinolone esters. Ann Trop Med Parasitol. 1970;64:209–22.
Muregi FW, Ohta I, Masato U, Kino H, Ishih A. Resistance of a rodent malaria parasite to a thymidylate synthase inhibitor induces an apoptotic parasite death and imposes a huge cost of fitness. PLoS ONE. 2011;6:e21251.
Da Silva Barbosa A, Ferreira MES, dos Santos Carmo L, Green MD, Dolabela MF, Percario S. Evans blue staining for the assessment of blood–brain barrier integrity in experimental malaria. Eur J Biomed Res. 2016;2:15–7.
Schlede E, Genschow E, Spielmann H, Stropp G, Kayser D. Oral acute toxic class method: a successful alternative to the oral LD 50 test. Regul Toxicol Pharmacol. 2005;42:15–23.
Kelly JX, Smilkstein MJ, Cooper RA, Lane KD, Johnson RA, Janowsky A, et al. Design, synthesis, and evaluation of 10-N-substituted acridones as novel chemosensitizers in Plasmodium falciparum. Antimicrob Agents Chemother. 2007;51:4133–40.
Nacer A, Movila A, Sohet F, Girgis NM, Gundra UM, Loke P, et al. Experimental cerebral malaria pathogenesis—hemodynamics at the blood brain barrier. PLoS Pathog. 2014;10:e1004528.
Baptista FG, Pamplona A, Pena AC, Mota MM, Pied S, Vigario AM. Accumulation of Plasmodium berghei-infected red blood cells in the brain are crucial for the development of cerebral malaria in mice. Infect Immun. 2010;78:4033–9.
Hayano M, Kojima S, Hanum S. Cytokine and chemokine responses in a cerebral malaria-susceptible or -resistant strain of mice to Plasmodium berghei ANKA infection: early chemokine expression in the brain. Int Immunol. 2003;15:633–40.
Clemmer L, Martins YC, Zanini GM, Frangos JA, Carvalho LJ. Artemether and artesunate show the highest efficacies in rescuing mice with late-stage cerebral malaria and rapidly decrease leukocyte accumulation in the brain. Antimicrob Agents Chemother. 2011;55:1383–90.
Carvalho LJ. Murine cerebral malaria: how far from human cerebral malaria? Trends Parasitol. 2010;26:271–2.
Berendt AR, Tumer GDH, Newbold CI. Cerebral malaria: the sequestration hypothesis. Parasitol Today. 1994;10:412–4.
Rénia L, Howland SW, Claser C, Gruner AC, Suwanarusk R, Hui T, et al. Cerebral malaria: mysteries at the blood–brain barrier. Virulence. 2012;3:193–201.
Wassmer SC, Moxon CA, Taylor T, Grau GE, Molyneux ME, Craig AG. Vascular endothelial cells cultured from patients with cerebral or uncomplicated malaria exhibit differential reactivity to TNF. Cell Microbiol. 2011;13:198–209.
Coltel N, Combes V, Hunt NH, Grau GE. Cerebral malaria—a neurovascular pathology with many riddles still to be solved. Curr Neurovasc Res. 2004;1:91–110.
Abbott NJ, Rönnbäck L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53.
Luissint A-C, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9:23.
Medana IM, Turner GD. Human cerebral malaria and the blood–brain barrier. Int J Parasitol. 2006;36:555–68.
Morrell CN, Srivastava K, Swaim A, Lee MT, Chen J, Nagineni C, et al. Beta interferon suppresses the development of experimental cerebral malaria. Infect Immun. 2011;79:1750–8.
Herbas MS, Okazaki M, Terao E, Xuan X, Arai H, Suzuki H. α-Tocopherol transfer protein inhibition is effective in the prevention of cerebral malaria in mice. Am J Clin Nutr. 2010;91:200–7.
Tamura T, Kimura K, Yuda M, Yui K. Prevention of experimental cerebral malaria by Flt3 ligand during infection with Plasmodium berghei ANKA. Infect Immun. 2011;79:3947–56.
Authors’ contributions
OCO, HNW, BJM, GNM, PGK, and FWM were involved in the study conception and design. FWM developed the original concept of the study. HNW synthesized the trioxaquine. OCO carried out the laboratory procedures, generated the tables, figures and wrote the initial draft. OCO, BJM and FWM scrutinized, interpreted the results, and had primary responsibility for final content. All authors read and approved the final manuscript.
Acknowledgements
Our gratitude goes to the Animal House staff of KEMRI, led by Mr. Lucas Ogutu, for their assistance during mice breeding.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Ethics statement
The use of mice in this research work was approved by both Animal Care and Use Committee (ACUC), and Scientific Ethical Review Unit (SERU) of KEMRI, Approval No. KEMRI/ACUC/01.04.16, and Permit No. KEMRI/RES/7/3/1/2016), respectively. The two units are accredited by the National Commission for Science, Technology and Innovation (NACOSTI).
Funding
The authors are grateful to the World Academy of Sciences for the Advancement of Science in Developing Countries (TWAS, Grant Award Number: 11-082 RG/BIO/AF/AC_I; UNESCO FR: 3240262705) awarded to Prof. Francis W. Muregi.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Odhiambo, O.C., Wamakima, H.N., Magoma, G.N. et al. Efficacy and safety evaluation of a novel trioxaquine in the management of cerebral malaria in a mouse model. Malar J 16, 268 (2017). https://doi.org/10.1186/s12936-017-1917-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-017-1917-6