
Abstract
Background
The aim of this study was to identify prognostic long non-coding RNAs (lncRNAs) and develop a multi-lncRNA signature for suvival prediction in esophageal squamous cell carcinoma (ESCC).
Methods
The clinical and gene expression data from Gene Expression Omnibus database (GSE53624, n = 119) were obtianed as training set. A total of 98 paired ESCC tumor and normal tissues were detected by RNA sequencing and used as test set. Another 84 ESCC tissues were used for real-time quantitative PCR(qRT-PCR) and as an independent validation cohort. Survival analysis, Cox regression and Kaplan–Meier analysis were performed.
Results
We screened a prognostic marker of ESCC from the GSE53624 dataset and named it as the five-lncRNA signature including AC007179.1, MORF4L2-AS1, RP11-488I20.9, RP13-30A9.2, RP4-735C1.6, which could classify patients into high- and low-risk groups with significantly different survival(median survival: 1.75 years vs. 4.01 years, log rank P < 0.05). Then test dataset and validation dataset confirmed that the five-lncRNA signature can determine the prognosis of ESCC patients. Predictive independence of the prognostic marker was proved by multivariable Cox regression analyses in the three datasets (P < 0.05). In addition, the signature was found to be better than TNM stage in terms of prognosis.
Conclusion
The five-lncRNA signature could be a good prognostic biomarker for ESCC patients and has important clinical value.
Similar content being viewed by others

Background
Esophageal carcinoma is a malignant tumor of esophageal mucosa epithelium or gland. Globally, esophageal cancer ranks seventh in morbidity and sixth mortality among all cancers [1]. China is one of the countries with the highest incidence of esophageal cancer, accounting for approximately half of all esophageal cancer cases [2]. There were 477,900 new cases diagnosed in China in 2015 [3]. Among these Chinese esophageal cancer patients, esophageal squamous cell carcinoma (ESCC) patients account for nearly 90% [2]. In terms of mortality, esophageal cancer deaths in China account for half of global esophageal cancer deaths [4]. Therefore, ESCC is a major public health challenge in the world, especially in China. Using Chinese ESCC patients as research samples is of great significance in revealing the pathogenesis and prognostic factors of ESCC.
ESCC is an extremely aggressive malignancy. Along with the clinical symptoms of early esophageal squamous cell carcinoma are not obvious and can be easily ignored by patients, most ESCC patients are diagnosed in advanced stages. As a result, the prognosis of ESCC patients is extremely poor. The 5 year survival rate of ESCC patients in some less developed areas is less than 10% [5]. In the United States, where medical conditions are advanced, the 5 year survival rate is 18% [6]. In China, the 5 year relative survival of postoperative ESCC patients is 8%-41% [7, 8]. Despite recent advances in diagnostic and therapeutic approaches, the life expectancy of ESCC patients has not improved significantly [3, 9, 10]. In addition, the current global diagnosis of ESCC depends on endoscopy and biopsy [11]. After confirming the diagnosis, TNM stage is used clinically to make prognostic judgment and guide treatment, but the shortcoming of TNM stage is that it cannot achieve individualized prediction and accurate evaluation [12, 13]. Therefore, identification of a sensitive and effective prognostic marker of ESCC is in urgent need to evaluate disease progression and patient’s overall outcome.
Next generation sequencing (NGS) and bioinformatics analysis tools provides great help for human-beings to understand tumors [14,15,16,17]. In the process of exploring tumor pathogenesis and prognostic factors through NGS and bioinformatics analysis, long non-coding RNAs (lncRNAs), defined as transcripts longer than 200 bp, have been discovered to play important roles. For example, a HOX transcript antisense RNA, lncRNA HOTAIR, was found to correlate with poor prognosis of lung cancer and promote tumor progression [18]. LncRNA REG1CP was shown to promote the development and progression of tumor [19]. For ESCC, several lncRNAs have been detected to associate with short survival, such as lncRNA H19 [20], lncRNA CASC9 [21] and lncRNA LUCAT1 [22]. Subsequently, prognostic multi-lncRNA signatures with high potential clinical application significance were constructed based on lncRNA expression profile data from public database such as Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) database. For instance, a three-lncRNA signature (ENST00000435885.1, XLOC_013014, ENST00000547963.1) was found that it can classify the ESCC patients into two groups with significantly different overall survival [23]. A seven-lncRNA (RP5-1172N10.2, RP11-579D7.4, RP11-89N17.4, LA16c-325D7.2, RP1-251M9.2, RP11-259O2.2, LINC00173) signature can predict overall survival of ESCC patients [24]. Although prognostic lncRNA signatures have been identified for ESCC, there are few studies that can verify the effectiveness of the predictive model in independent experimental data set and confirm the value of the prognostic lncRNA in ESCC tissues.
Here, we used our lncRNA expression data tested by RNA sequencing and followed up the clinical 5 year survival information of 98 ESCC patients, then, combined it with 119 ESCC public expression profiles from GEO and another independent 84 ESCC tissues for qPCR validation to construct a clinically valuable lncRNA signature that can accurately predict survival of ESCC patients.
Materials and methods
Sample collection and preparation
LncRNA expression profile and corresponding clinical data of 119 ESCC cases (GSE53624) were obtained from the publicly available GEO database (https://www.ncbi.nlm.nih.gov/geo/). As Anyang is a high-risk area of ESCC in China, we collected 98 postoperative ESCC tissues and paired non-tumor tissues from Anyang Tumor Hospital during 2014–2015, and organized relevant patient clinical information. Then we examined the protein coding gene (PCG) and lncRNA expression profile of the 98 pair ESCC tissues by next-generation sequencing (NGS, Hereinafter referred to as RNA-seq dataset). In addition, we collected an independent validation cohort including 84 postoperative ESCC patients from the same hospital and detected their lncRNA expression level using the qRT-PCR. Detailed clinicopathological characteristics of all these ESCC patients of the ESCCs in this study was shown in Table 1. Tumor-node-metastasis (TNM) classification of the International Union against Cancer, 7th edition was used to categorize. Documentation of informed consent was obtained through the institutional review board. The study was approved by the Anyang Tumor Hospital Ethical Committee.
RNA isolation and next generation RNA sequencing analysis
After TRIZOL lysis and purification, total RNA was isolated by the miRNeasy Mini Kit (QIAGEN) with DNase digestion step. A total amount of 5 ug RNA per sample was used as input material for the RNA sample preparation. Firstly, ribosomal RNA was removed by Epicentre Ribozero™ rRNA Removal Kit (Epicentre, USA), and rRNA free residue was cleaned up by ethanol precipitation. The sequencing libraries were generated by NEBNext Ultra™ Directional RNA Library Prep Kit for Illumina (NEB, USA) following manufacturer's recommendations and sequenced on an Illumina Hiseq platform. The 150 bp paired-end reads were generated. Normalized fragments per kilobase per million mapped reads (FPKM) by cufflinks was used to estimate the gene expression values.
Validation of lncRNA expression using RT-PCR
LncRNAs reverse transcription were amplified with TIANScript II RT Kit (KR107, TIANGEN, Beijing, China). We used real-time quantitative PCR (qRT-PCR) to measure the lncRNA expression with TB Green® Premix Ex Taq™ (Tli RNaseH Plus, TaKaRa, Dalian, China). Relative quantification of lncRNA expression was normalized by the 2−ΔCt method, and GAPDH was used for normalization with the corresponding primers (Additional file 1. Table S1). All reactions were carried out in triplicate by StepOnePlus™ Real-Time PCR System (Applied Biosystems) [25,26,27].
Construction of multi-lncRNA prognostic signature
GSE53624 and RNA-seq datasets were analyzed respectively by Cox analysis and Kaplan–Meier analysis to identify lncRNAs associated with overall survival (OS) of ESCC patients. We selected the prognostic lncRNAs using Cox p & log rank P < 0.05 in 2 datasets. Model was estimated as follows [25, 26, 28]: Risk Score (RS) = ∑ Ni=1 (ExpLncRNAi * CoefCoxi), where N is prognostic lncRNA number, ExpLncRNAi represents the lncRNA expression value, and CoefCoxi is the Cox regression coefficient of lncRNA. We plotted ROC curves [16] and calculated their area under the curve (AUC) values to screen out the prognostic signature with the largest AUC value in the GSE53624 set [28]. The whole construction process was shown in Additional file 2. Figure S1. R program (Version 3.5.1) was used to perform the above analyses, including packages called pROC, TimeROC and survival from Bio-conductor (https://www.bioconductor.org/). SubpathwayMiner was used to find the selected prognostic genes related pathways (https://cran.r-project.org/web/packages/SubpathwayMiner/), which provides more flexibility in annotating gene sets and identifying the involved pathways (entire pathways and sub-pathways) [29].
Results
Screening prognostic lncRNAs in ESCC
There were a total of 217 samples with lncRNA expression profiles including GSE53624 (n = 119) and RNA-seq (n = 98) dataset in this study. From the above two datasets, we found a total of 6253 lncRNAs were expressed in ESCC tissues. We then performed univariate cox analysis and Kaplan–Meier analysis to analyze the relationship between the lncRNA expression and ESCC OS. Based on the two ESCC profiling datasets and the corresponding clinical follow-up information, univariate cox analyses identified 368 lncRNAs in the GSE53624 and 290 lncRNAs in the RNA-seq dataset which were significantly associated with ESCC OS (Cox P < 0.05) and Kaplan–Meier analyses identified 386 survival associated lncRNAs in the GSE53624 and 312 survival associated lncRNAs in the RNA-seq dataset (log rank P < 0.05). Through comparing the above four groups of survival related lncRNA data, we found 19 identical lncRNAs significantly correlated with OS in two datasets (COX P < 0.05, log rank P < 0.05) (Fig. 1a). Subsequently, we found that 10 of the 19 prognostic-related lncRNAs showed a consistent risk trend in the four groups, displaying a risk or protective role in ESCC (Additional file 3. Table S2).

Constructing the prognostic lncRNA model in the high throughput sequencing datasets. a Venn diagram for analyzing the prognostic lncRNAs. b Screening out the lncRNA signature with largest AUC from all 1023 signatures which were calculated by ROC for k = 1, 2…… 10. c The AUC of the screened lncRNA signature
Constructing prognostic multi-lncRNA signatures
We used 119 ESCC data from public GSE53624 as the training group, 98 RNA-seq data as test group and 84 qPCR data as validation group. Using above selected 10 prognostic lncRNAs, we constructed 210–1 = 1023 signatures in the training dataset. In order to obtain a signature with the strongest prognostic ability, we performed ROC analyses in the training dataset (Additional file 4. Table S3). We screened the 5-lncRNA combination with the largest AUC value, namely the prognostic signature containing AC007179.1, MORF4L2-AS1, RP11-488I20.9, RP13-30A9.2, RP4-735C1.6 (Fig. 1b, Table 2). The lncRNA risk score was calculated as follows: (0.47 × expression value of AC007179.1) + (0.58 × expression value of MORF4L2-AS1) + (-0.47 × expression value of RP11-488I20.9) + (-0.64 × expression value of RP13-30A9.2) + (0.62 × expression value of RP4-735C1.6). As shown in Fig. 1c, the AUC of the screened lncRNA signature was 0.71.
Predictive power of the lncRNA signature for patients with ESCC
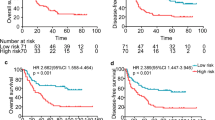
After calculated risk scores of ESCC patients in the GSE53624 dataset, the median risk classified ESCC patients into high- and low- risk groups (n = 59/60). Kaplan–Meier analysis found the survival of ESCC patients in the low-risk group was significantly longer than those in the high-risk group (median survival: 1.75 years vs. 4.01 years, log-rank test P < 0.001; Fig. 2a). The 3 year survival rate for patients in the high-risk group was only 23.73%, while that of patients in the low-risk group reached as high as 66.67%.

The lncRNA signature classification power for ESCC prognosis. Kaplan–Meier curves found ESCC patients were classified into two different risk groups based on the risk score of the signature in the GSE53624 (a), RNA-seq (b) and qPCR validation datasets (c)
Then we evaluated the predictive ability of the five-lncRNA signature in ESCC RNA-seq dataset (n = 98), which was treated as the test set. The median risk score of patients in the test dataset was calculated and used to divide patients into high- and low-risk groups. Figure 2b showed the Kaplan–Meier analysis result. The prognosis of ESCC patients in the low-risk group was significantly better than patients in the high-risk group (Median survival: 2.44 years vs. 3.97 years, log-rank test P = 0.014; Fig. 2b). The 3 year survival rate for the patients of high-risk group was only 40.82%, while that of the low-risk group was 61.22%.
In addition, we performed qPCR experiment and obtained the five lncRNAs expression level in 84 ESCC tissues. Through calculated risk scores and used the median risk score as the cutoff value, we obtained the high score group and low score group. Kaplan–Meier analysis discovered the survival difference between two risk groups. The survival of ESCC patients with low-risk scores was still significantly greater than patients with high-risk scores. (Median survival: 2.82 years vs. 4.08 years, log-rank test P = 0.011; Fig. 2c).
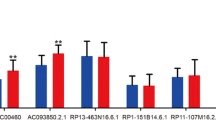
Figure 3 displayed the survival time, risk score and lncRNA expression level of each patient in above two ESCC lncRNA expression profile datasets, including GSE53624 and RNA-seq datasets. As shown, patients with the higher expression of AC007179.1, MORF4L2-AS1 and RP4-735C1.6, the higher the risk were with the more deaths. On the contrary, patients with lower-risk scores tended to have higher expression level of protective lncRNAs (RP13-30A9.2, RP11-488I20.9) and have a lower mortality rate.

Risk score distribution, survival status and gene expression of ESCC patients in high- and low-risk groups classified by the four-lncRNA signature in the GSE53624 (a), RNA-seq (b) datasets
The five-lncRNA signature can independently predict the survival of ESCC patients
The Chi-square test found the lncRNA signature was not associated with clinicopathological factors, including age, sex and pTNM stage in the training, test and qPCR validation groups (Table 3). Although univariate cox analysis of the training, test and independent validation datasets showed TNM stage and the signature were both associated with the prognosis of ESCC patients, multivariable Cox regression analyses showed only the lncRNA signature was an independent prognostic factor for ESCC patients (High-risk group vs. Low-risk group, HR training = 3.50, 95% CI 2.13–5.76, P < 0.001, n = 119; HR test = 1.03, 95% CI 1.00–1.06, P = 0.014, n = 98; HR independent = 2.95, 95% CI 1.21–7.17, P = 0.017, n = 84, Table 4).
The prediction performance of the five-lncRNA signature and TNM stage
TNM stage is currently used as the main indicator for prognosis evaluation of ESCC. Our study confirmed that TNM stage and the five-lncRNA signature were related to the prognosis of patients. Therefore, we performed stratification analysis to explore the predictive power of the five-lncRNA signature in patients with known TNM stage. We first divided all the ESCC patients involved in this study (n = 301) into 2 groups, one group was patients with TNM low (I + II) stage and the other group was patients with high TNM stage (III + IV). Then we used the lncRNA signature to conduct stratification analyses of above two groups. Kaplan–Meier results showed that the lncRNA signature could further separate the patients with TNM low (I + II) stage (Fig. 4a) or patients with high TNM stage (III + IV)(Fig. 4b) into two subgroups with significantly different survival(log-rank test P < 0.001).

Stratification analysis of TNM stage using the five-lncRNA signature. a, b Stratification analysis of TNM low/high stage using the signature by Kaplan–Meier curves. c, d Comparing the survival prediction power of the signature with that of TNM stage by ROC in the entire datasets. e TimeROC analysis to explore the survival prediction power of combination of the signature and TNM stage in the entire dataset
To analyze the predictive performance advantages of the five-lncRNA signature, TimeROC analyses were performed in the three datasets (n = 301). The AUCs of the signature were 0.690/0.648/0.639 at 5/3/1 years (Fig. 4c), while the AUCs of TNM were 0.643/0.654/0.637 at 5/3/1 years (Fig. 4d), suggesting the five-lncRNA signature was superior to TNM stage in terms of ESCC prognosis evaluation, especially at 5 years survival. Moreover, we found AUC values were maximized when signature and TNM staging were combined to assess patient prognosis (0.742/0.723/0.702 at 5/3/1 years, Fig. 4e), indicating the lncRNA signature improve the accuracy of TNM in evaluating prognosis.
Functional prediction of the five-lncRNAs signature
In order to predict function of the five-lncRNAs signature, we performed Pearson analysis to obtain the genes implicated in the correlation of these 5-lncRNA signature in the GSE53624 and RNAseq datasets, respectively. Then we got 988 co-expression genes in total (Pearson coefficient > 0.4/ < -0.4, P < 0.001, Fig. 5a). SubpathwayMiner suggested these 988 genes were significantly enriched in 73 different KEGG pathways (P < 0.05, Additional file 5. Table S4), especially involved in p53 signaling pathway, ErbB signaling pathway, Regulation of actin cytoskeleton, MAPK signaling pathway, PPAR signaling pathway VEGF signaling pathway, Pathways in cancer and Toll-like receptor signaling pathway (Fig. 5b), which were ranked in the top 20 and were closely related to the cancer development.

Functional prediction of the five-lncRNAs signature. Visualization of the co-expressing genes with the risk score signature in GS53624 and RNA-Seq datasets (a). Functional enrichment analysis of those co-expressing genes by SubpathwayMiner (b)
Discussion
Esophageal squamous cell carcinoma is a rapidly progressive disease faced with many difficulties, such as difficulty in early diagnosis, low 5 year survival rate, and lack of effective prognostic markers. Recently, lncRNA has been reported to be involved in the occurrence and progression of tumors [30, 31]. However, the expression characteristics and roles of lncRNAs in ESCC are still fairly elusive. Thus, this paper revealed the survival related lncRNAs expression level in ESCC and constructed a prognostic five-lncRNA signature by collected public ESCC lncRNA expression data. Then, we measured the lncRNA expression of 98 ESCC patients by RNA seq and test the lncRNA expression of 84 ESCC tissues for validating the predictive performance of the five-lncRNA signature. Meanwhile, the 98 ESCC lncRNA expression profiles provide scientists with reference data to study the association of ESCC and lncRNAs.
Despite numerous research have reported some ESCC lncRNA models for prognostic markers [24], PCR validation was rarely performed in the independent validation group and some lncRNAs in the prediction model might not yet exist in ESCC tissue. In this study, the predictive model was constructed through the analyzing two sets of high-throughput sequencing data, and lncRNAs of the five-lncRNA signature were shown to be present in collected ESCC samples by qPCR.
In the five-lncRNA suvival prediction model, the high expression level of lncRNA MORF4L2-AS1, AC007179.1 and RP4-735C1.6 were associated with short survival (Cox coefficient > 0), indicating these three lncRNAs were risk lncRNAs for ESCC patients. The high expression of RP13-30A9.2 and RP11-488I20.9 were associated with long survival (Cox coefficient < 0), suggesting the two lncRNAs were protective lncRNAs for ESCC. The significant correlation of the five lncRNAs with ESCC prognosis was found in two data sets, emphasizing the important clinical research value of the five prognostic lncRNAs in ESCC. However, the function of prognostic lncRNAs in ESCC have not been reported so far and biological roles of the five lncRNAs in ESCC progression should be investigated in further experimental studies.
TNM stage is a commonly used tumor classification standard in clinical practice and a recognized prognostic marker [32, 33]. However, TNM stage remains flawed in prognostic assessment. We found that the prognostic predictive power of signature is better than TNM stage, suggesting that the strong prognostic ability of the five-lncRNA signature. Consistent with some scholars’ results that combined TNM classification with molecular marker can predict outcome of ESCC patients more accurately [34], we found the prognostic prediction of the combination of signature with TNM stage was the best, indicating the signature combined with TNM stageg is useful for prognosis evaluation.
Conclusion
We identified a prognostic five-lncRNA signature from a large cohort of ESCC patients. The signature could predict the survival of patients with ESCC based on lncRNA expression profile and have strong clinical value.
Availability of data and materials
The data generated during this study are available from the corresponding author upon reasonable request. The data that support the findings of this study are available in Gene Expression Omnibus database (GEO). These data were derived from the following resources available in the public domain: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53624.
Abbreviations
- ESCC:
-
Esophageal squamous cell carcinoma
- qPCR:
-
Real-time quantitative PCR
- ROC:
-
Receiver operating characteristic
- AUC:
-
Area under the ROC curve
- CI:
-
Confidence interval
- OS:
-
Overall survival
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Malhotra GK, Yanala U, Ravipati A, Follet M, Vijayakumar M, Are C. Global trends in esophageal cancer. J Surg Oncol. 2017;115(5):564–79.
Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–32.
Kou K, Guo X, Baade P, Lu Z, Fu Z, Chu J, Xu A, Sun J. Spatial analysis of esophageal cancer mortality in a high-risk population in China: consistent clustering pattern in 1970–74 and 2011–13. Asian Pac J Cancer Prev. 2018;19(11):3161–6.
Aghcheli K, Marjani HA, Nasrollahzadeh D, Islami F, Shakeri R, Sotoudeh M, Abedi-Ardekani B, Ghavamnasiri MR, Razaei E, Khalilipour E, et al. Prognostic factors for esophageal squamous cell carcinoma–a population-based study in Golestan Province, Iran, a high incidence area. PLoS ONE. 2011;6(7):e22152.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29.
Li XM, Tang S, Peng XQ. Changes of five-year survival rates in patients with esophageal squamous cell#br# carcinoma from 1973 to 2005. Cancer Res Prev Treat. 2014;41(03):237–42.
Liu J, Xie X, Zhou C, Peng S, Rao D, Fu J. Which factors are associated with actual 5-year survival of oesophageal squamous cell carcinoma? Eur J Cardiothorac Surg. 2012;41(3):e7–11.
Zeng H, Zheng R, Zhang S, Zuo T, Xia C, Zou X, Chen W. Esophageal cancer statistics in China, 2011: estimates based on 177 cancer registries. Thorac Cancer. 2016;7(2):232–7.
Patel N, Benipal B. Incidence of esophageal cancer in the United States from 2001–2015: a United States cancer statistics analysis of 50 states. Cureus. 2018;10(12):e3709.
Lagergren J, Smyth E, Cunningham D, Lagergren P. Oesophageal cancer. Lancet. 2017;390(10110):2383–96.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol. 2010;17(6):1471–4.
Tripathy D, Harnden K, Blackwell K, Robson M. Next generation sequencing and tumor mutation profiling: are we ready for routine use in the oncology clinic? BMC Med. 2014;12:140.
Biesecker LG, Burke W, Kohane I, Plon SE, Zimmern R. Next-generation sequencing in the clinic: are we ready? Nat Rev Genet. 2012;13(11):818–24.
Guo JC, Fang SS, Wu Y, Zhang JH, Chen Y, Liu J, Wu B, Wu JR, Li EM, Xu LY, et al. CNIT: a fast and accurate web tool for identifying protein-coding and long non-coding transcripts based on intrinsic sequence composition. Nucleic Acids Res. 2019;47(W1):W516–W522522.
Wu Y, Zhang F, Yang K, Fang S, Bu D, Li H, Sun L, Hu H, Gao K, Wang W, et al. SymMap: an integrative database of traditional Chinese medicine enhanced by symptom mapping. Nucleic Acids Res. 2019;47(D1):D1110–D111717.
Loewen G, Jayawickramarajah J, Zhuo Y, Shan B. Functions of lncRNA HOTAIR in lung cancer. J Hematol Oncol. 2014;7:90.
Yari H, Jin L, Teng L, Wang Y, Wu Y, Liu GZ, Gao W, Liang J, Xi Y, Feng YC, et al. LncRNA REG1CP promotes tumorigenesis through an enhancer complex to recruit FANCJ helicase for REG3A transcription. Nat Commun. 2019;10(1):5334.
Li X, Yang H, Wang J, Li X, Fan Z, Zhao J, Liu L, Zhang M, Goscinski MA, Wang J, et al. High level of lncRNA H19 expression is associated with shorter survival in esophageal squamous cell cancer patients. Pathol Res Pract. 2019;215(11):152638.
Liang Y, Chen X, Wu Y, Li J, Zhang S, Wang K, Guan X, Yang K, Bai Y. LncRNA CASC9 promotes esophageal squamous cell carcinoma metastasis through upregulating LAMC2 expression by interacting with the CREB-binding protein. Cell Death Differ. 2018;25(11):1980–95.
Yoon JH, You BH, Park CH, Kim YJ, Nam JW, Lee SK. The long noncoding RNA LUCAT1 promotes tumorigenesis by controlling ubiquitination and stability of DNA methyltransferase 1 in esophageal squamous cell carcinoma. Cancer Lett. 2018;417:47–57.
Li J, Chen Z, Tian L, Zhou C, He MY, Gao Y, Wang S, Zhou F, Shi S, Feng X, et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut. 2014;63(11):1700–10.
Mao Y, Fu Z, Zhang Y, Dong L, Zhang Y, Zhang Q, Li X, Liu J. A seven-lncRNA signature predicts overall survival in esophageal squamous cell carcinoma. Sci Rep. 2018;8(1):8823.
Guo JC, Xie YM, Ran LQ, Cao HH, Sun C, Wu JY, Wu ZY, Liao LD, Zhao WJ, Fang WK, et al. L1CAM drives oncogenicity in esophageal squamous cell carcinoma by stimulation of ezrin transcription. J Mol Med. 2017;95(12):1355–68.
Guo JC, Li CQ, Wang QY, Zhao JM, Ding JY, Li EM, Xu LY. Protein-coding genes combined with long non-coding RNAs predict prognosis in esophageal squamous cell carcinoma patients as a novel clinical multi-dimensional signature. Mol BioSyst. 2016;12(11):3467–77.
Zhang XD, Huang GW, Xie YH, He JZ, Guo JC, Xu XE, Liao LD, Xie YM, Song YM, Li EM, et al. The interaction of lncRNA EZR-AS1 with SMYD3 maintains overexpression of EZR in ESCC cells. Nucleic Acids Res. 2018;46(4):1793–809.
Guo JC, Wu Y, Chen Y, Pan F, Wu ZY, Zhang JS, Wu JY, Xu XE, Zhao JM, Li EM, et al. Protein-coding genes combined with long noncoding RNA as a novel transcriptome molecular staging model to predict the survival of patients with esophageal squamous cell carcinoma. Cancer Commun. 2018;38(1):4.
Li C, Li X, Miao Y, Wang Q, Jiang W, Xu C, Li J, Han J, Zhang F, Gong B, et al. SubpathwayMiner: a software package for flexible identification of pathways. Nucleic Acids Res. 2009;37(19):e131.
Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Can Res. 2017;77(15):3965–81.
Slack FJ, Chinnaiyan AM. The Role of non-coding RNAs in oncology. Cell. 2019;179(5):1033–55.
Sobin LH, Fleming ID. TNM classification of malignant tumors, fifth edition (1997). Union Internationale Contre le Cancer and the American Joint Committee on Cancer. Cancer. 1997;80(9):1803–4.
Sobin LH, Hermanek P, Hutter RV. TNM classification of malignant tumors. A comparison between the new (1987) and the old editions. Cancer. 1988;61(11):2310–4.
Takeno S, Noguchi T, Takahashi Y, Fumoto S, Shibata T, Kawahara K. Assessment of clinical outcome in patients with esophageal squamous cell carcinoma using TNM classification score and molecular biological classification. Ann Surg Oncol. 2007;14(4):1431–8.
Acknowledgements
The authors gratefully thank the academic editor and the anonymous reviewers for their insightful comments and suggestions to improve this manuscript.
Funding
This work was supported by grant from National Natural Science Foundation of China (No. 81272371 and No. U1904148), the National Science and Technology Major Project of China (No. 2018ZX10302205), Zhengzhou Major Project for Collaborative Innovation (Zhengzhou University, No. 18XTZX12007), Scientific and technological project in Henan Province (No. 192102310057), and the Youth Innovation Fund of The First Affiliated Hospital of Zhengzhou University to Wei-Wei Wang and Pan Li.
Author information
Authors and Affiliations
Contributions
The authors contributed in the following way: LZ: data collection, data analysis, interpretation, and drafting; GJ, WW: study design, study supervision, and final approval of the manuscript; PL, EL, CX, DZ, JZ: technical support, experiment advice and data collection. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
The detail of lncRNAs and GAPDH primers in this study.
Additional file 2: Figure S1.
The schedule of analyses to construct the lncRNA signature in this study.
Additional file 3: Table S2.
Ten out of the 19 prognostic-related lncRNAs showed a consistent risk trend in the high throughput sequencing data.
Additional file 4: Table S3.
The 1023 signatures comprising lncRNAs in the GSE53624 dataset (n = 119)
Additional file 5: Table S4.
Functional enrichment of those co-expressed genes with the risk score by SubpathwayMiner analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
zhang, L., Li, P., Liu, E. et al. Prognostic value of a five-lncRNA signature in esophageal squamous cell carcinoma. Cancer Cell Int 20, 386 (2020). https://doi.org/10.1186/s12935-020-01480-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01480-9