
Abstract
N6-methyl-adenosine(m6A) modification emerges as an abundant and dynamic regulation throughout the Eukaryotic transcriptome. Dysregulation of the m6A regulators has increasingly been found in many neoplasms. It is reasonable to believe that m6A changes the fate of cancer cells and subsequently affected all aspects of cancer progression. In view of the context-dependent role of m6A modification, we emphasize a dual effect of m6A in a particular tumor model, that is, m6A plays a promoting role or a suppressing role in different stages of cancer. This novel sight is compared to the older view that a particular m6A regulator acts as a consistent role in cancer progression.
Similar content being viewed by others
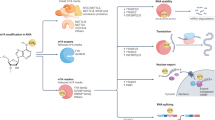

Background
m6A modification is the most abundant eukaryotic RNA modification and can regulate many essential biological processes of the transcriptome, such as RNA splicing, nuclear export, stability, translation, and decay. m6A modification in human tumor cells has a significant influence on tumorigenesis, stemness, proliferation, invasion, metastasis, and response to immune via regulation of many oncogenes and tumor suppressor genes. Therefore, control of m6A modification in tumor cells is a potential anticancer therapeutic target.
m6A was first detected in a poly(A)-mRNA fraction as a mode of mRNA chemical modification regulation in 1974 [1]. However, there has been no gratifying progress on the study of m6A due to lack of methods for detecting m6A sites in transcriptome. Researchers were not able to identify the individual m6A-RNA site and it was tough to distinguish m6A from adenosine (A) or 2′-O-methyl-N6-methyl-Adenosine Ribonucleic Acid (m6Am) (Fig. 1). m6Am is able to react with m6A-specific antibody m6Am contains the same N6-methyl as m6A on the basis of 2′-O-methyl-adonesine.

Chemical structure of adenosine modification
The innovation and development of high-throughput sequencing have significantly improved the above situation. By combining highly specific m6A antibodies and high-throughput sequencing, two scientific groups independently developed the first transcriptome-wide m6A sites mapping method termed Methylated RNA Immunoprecipitation Sequencing (MeRIP-Seq) in individual RNAs [2, 3]. This method could identify tens of thousands of candidate m6A modification sites at an average resolution of 100 nucleotides. Liu et al. developed a low-throughput method termed Site-specific Cleavage And Radioactive-labeling followed by Ligation-assisted Extraction and Thin-layer chromatography (SCARLET) that could detect the presence of the particular modification fractions [4]. In addition, m6A individual-nucleotide-resolution Crosslinking and Immunoprecipitation (miCLIP) makes single-nucleotide-resolution mapping possible [5].
These technological advances have revealed many novel characteristics and mechanisms of this pervasive transcriptome modification. Researchers surprisingly found that m6A is highly selective: [1] Only some of RNAs contain m6A [2]; 2 m6A usually appears near stop codon and in 3′UTR [3]; 3 The DRACH (D = G, A, or U; R = G or A; and H = C, A, or U) motif is a consensus sequence of the m6A modification [6, 7]. The level of m6A in transcriptome was determined to be dynamic, varying in cell development and response to stresses [2, 3].
The idea that adenosine N6-methylation is reversible provides the foundation of the m6A dynamic regulation hypothesis. N6 sites of RNA adenosine can be methylated by special methyltransferase, such as Methyltransferase-like 3 (METTL3) [8, 9]. Various kinds of RNA binding proteins exist in Eukaryotic cells. Some of them can recognize and decode m6A, then change the structures of modified RNAs, and finally determine the fates of m6A-RNAs. Among them, what we know most about is the YTH domain protein family [10]. In the past few years, other m6A binding proteins have been discovered. Some results of studies on how these m6A binding proteins work and what they do are controversial or even opposite. m6A can be removed by demethylase, like fat mass and obesity-associated protein (FTO) and Alpha-ketoglutarate-dependent dioxygenase (ALKB) homolog 5 (ALKBH5) [11, 12]. Controversies also exist regarding the effects of RNA demethylases, which we describe in detail below. Thus, ‘writer’ adding m6A modification, ‘reader’ decoding m6A modification and embodying its roles, and ‘eraser’ exerting demethylation activity form a network of m6A dynamic regulation (Fig. 2). Once the m6A “Reader-Writer-Eraser” model was proposed, it has become the core of the m6A study. However, we still know very little about the newly discovered m6A regulators, and many gaps lie in our understanding of the dynamic and selective regulation mechanism of m6A. Therefore, improved understanding of the m6A “Reader-Writer-Eraser” model and its working mechanism in human cancer should lead to novel anticancer strategy.

Simplified model of m6A Dynamic regulation
With the deepening understanding of m6A, it is hard to simply categorize an m6A regulator into an oncogene or tumor suppressor in different cancer models. For instance, the eraser FTO may act as an oncogene in lung carcinoma [13] and as a tumor suppressor in renal clear cell carcinoma [14]. Also certain protein may have dual effects in different progression stages of an exclusive tumor model. One example is that YTHDF2, an m6A reader, may promote the proliferation of pancreatic cancer cells, whereas it suppresses metastasis in pancreatic cancer [15]. Arguments on the role of m6A modification in transcriptome hinder the application of this important posttranscriptional regulation to cancer diagnosis and treatment, hence reshaping the role of m6A modification in human cancer becomes crucial.
This review is to provide the current view of m6A regulating transcriptome dynamically and selectively through the novel regulatory model, and facilitate a better understanding of the complex effects of m6A modification on the transcriptome of human cancer cells.
m6A writer
Rottman et al. first discovered and purified the first m6A methyltransferase, now referred to as METTL3 [8, 9]. Subsequently, other suspected methyltransferases were discovered, such as Methyltransferase-like 14 (METTL14) and Wilms’ tumor 1-associated protein (WTAP), although finally confirmed that they had no methyltransferase activity. The latest sight is that it is METTL3-METTL14-WTAP methyltransferase complex that is located to nuclear speckles and responsible for m6A formation.
S-adenosyl methionine (SAM) is an important auxiliary substrate involved in transmethylation. METTL3 is a member of SAM-dependent methyltransferase family, which widely exist in eukaryotic cells and is highly conserved in mammals [16]. Genetic deletion of METTL3 results in nearly complete loss of m6A in A-rich RNA [17,18,19]. Thus, METTL3 is the main m6A methyltransferase of eukaryotic poly-A mRNA. METTL3 does not contribute to m6A formation of rRNA and small nuclear RNA (snRNA) [20]. Therefore, novel m6A writers remain to be discovered to fill this gap.
Once it was discovered, METTL14 was regarded as the second methyltransferase enzyme [21]. However, three separate crystallization studies showed that METTL14 lacks a SAM-binding domain and that purified METTL14 does not have any methyltransferase activity [22,23,24]. Previous misunderstandings of data are considered to be due to METTL3 coprecipitation. Still, METTL14 does is instrumental in enhancing catalytic activity, as well as positioning the methyl group [22,23,24].
WTAP works as an adaptor protein without catalytic activity for m6A modification [25]. WTAP depletion causes loss of METTL3 and METTL14 in nuclear speckles and loss of m6A formation in mRNA. Thus, WTAP maintains METTL3 location. It is caused by the lack of the nuclear localization sequence (NLS) [26].
METTL3, METTL14, and WTAP form the m6A methyltransferase complex. METTL3, as the catalytic core, transfers the methyl group from the SAM to the RNA. METTL14 serves as the RNA-binding arm, promoting the affinity to the RNA substrate and enhancing the complex integrity [22, 27]. METTL3 and METTL14 form a stable asymmetric heterodimer, which then binds to WTAP [28]. WTAP assists m6A methyltransferase complex to localize in nuclear speckles [28].
m6A eraser
Ever since discovering m6A methyltransferase, people have been working hard to realize the idea of m6A dynamic regulation through searching enzymes that remove m6A methyl groups. Situation had been very upset until the discovery of the first m6A associated demethylase FTO in 2011 [11]. However, increasing evidences are pointing towards the idea that FTO acts on m6Am rather than m6A. Nevertheless, we will introduce it below. Another important demethylase is ALKBH5 [12]. Both FTO and ALKBH5 belong to the Fe2+-α-ketoglutarate-dependent dioxygenase family. Recent research found a new demethylase, ALKBH3, which prefers tRNA demethylation rather than mRNA or rRNA [29]. Considering the subtle effect of ALKBH5 on mRNA and m6A dynamic regulation of rRNA or other non-coding RNA, it is unsurprising that other unknown members of the ALKB family exist.
FTO
As FTO is able to demethylate the N6-position of A, FTO is the first enzyme associated with m6A demethylation. However, now, we prefer to believe that FTO is a demethylase for m6Am rather than m6A based on current findings. Firstly, several researches shook the connection between FTO and m6A. FTO did not show a preference for consensus site of m6A [11] whereas a strong preference for m6Am [30]. Additional indication was the low catalytic rate of FTO for m6A while its catalytic rate towards m6Am was much higher [30]. More importantly, FTO knockout brings on considerably increases in m6Am, with no detectable increase in m6A quantitative measurement. The counterpart is that the expression of FTO caused a reduction in m6Am, with no effect on m6A [30]. The detectable growth in m6A seen in FTO depletion may be attributed to a reactive rise of METTL14 [31]. Thus, FTO has been moving away from the core of m6A demethylation.
ALKBH5/3
In quantitative measurements for m6A in mRNA, ALKBH5 knockdown caused increases, and ALKBH5 overexpression resulted in decreases, supporting the idea that ALKBH5 might make a significant difference in demethylation of m6A in mRNA [12]. What’s more, unlike FTO, ALKBH5 has no activity towards m6Am and appears preference for m6A [30]. ALKBH5 knockout mice are mostly normal except for defects in spermatogenesis. Additionally, the effect of ALKBH5 to m6A is subtle [12]. Thus, the development of mammals doesn’t require m6A demethylation, unless an unknown m6A demethylase exists.
The research about ALKBH3 proved this prediction. Notably, ALKBH3 shows a preference for m6A in tRNA rather than in mRNA or other non-coding RNA. ALKBH3-mediated tRNA demethylation promotes translation efficiency in cancer cells [29].
m6A reader
The major mechanism by which m6A works is by recruiting m6A-binding proteins. Among them, the most comprehensive researches currently are about the YT521-B homology (YTH) domain-containing proteins family, including YTH-domain C1 (YTHDC1), YTH-domain C2 (YTHDC2), YTH-domain F1 (YTHDF1), YTH-domain F2 (YTHDF2) and YTH-domain F3 (YTHDF3). Recent years, many new m6A readers without YTH domain have been discovered. These readers, including eukaryotic initiation factor 3 (eIF3), Heterogeneous nuclear ribonucleoprotein family (HNRNPs), Proline rich coiled-coil 2A (Prrc2a) and Insulin-like growth factor 2 mRNA binding proteins (IGF2BPs), etc., have significantly enriched people’s understanding of the role of m6A modification in transcriptome.
YTH domain proteins
The YTH domain was initially identified by the homology search of a human splicing factor YT521B [32]. This 100–150 residues domain, which was detected to attribute to RNA-binding function of YT521B, forms a tryptophan cage comprising two or three tryptophan residues around the methyl group of m6A [33]. Then, Rechavi and colleagues first linked this structure to the m6A-binding protein. In the m6A RNA pull-down experiment, they detected that YTH domain YTHDF2 and YTHDF3 could bind to m6A [2]. Several subsequent research groups verified this result using gel shift assay and crystallography, and have inspired more exploration of the role and mechanism of these YTH domain proteins [7, 34, 35].
YTHDC1 is a nuclear protein involved in hnRNA splicing. YTHDC1 regulates exon inclusion by interacting with trans- and cis- regulatory elements, such as facilitating serine and arginine-rich splicing factor 3 (SRSF3) or blocking SRSF10 [36]. The long non-coding RNA (lncRNA) X-inactive specific transcript(XIST) is a m6A-riched lncRNA, which mediates X chromosome silencing. YTHDC1 preferentially recognizes m6A residues on XIST and promotes XIST function [37]. Moreover, recent studies have shown that YTHDC1 interacts with SRSF3 and nuclear RNA export factor 1 (NXF1) to promote the nuclear export of m6A-mRNAs [38]. Another research on RNA metabolism shows that recombinant Methionine Adenosyltransferase II Alpha (MAT2A) m6A is read by YTHDC1, which induces SAM-mediated MAT2A mRNA degradation [39].
YTHDC2 is a putative RNA helicase, aside from the YTH domain. YTHDC2 selectively binds to a few m6A sites, especially in non-coding RNA. Additionally, YTHDC2 can enhance translation efficiency and promote targeted RNA degradation by selectively binding m6A [40,41,42,43].
YTHDF1 ~ 3 are cytoplasmic m6A readers. It was initially demonstrated that YTHDF1 binds to the m6A site surrounding the stop codon and facilitate the translation initiation machinery to enhance the translation efficiency of targeted RNAs [44]. In contrast, YTHDF2 accelerates the degradation of m6A-modified RNA by directly recruiting CCR4-NOT deadenylase complexes [45]. YTHDF3 can be regarded as a fine-tuning of the RNA accessibility of YTHDF1 and YTHDF2 because YTHDF3 can synergize with YTHDF1 to promote the translation of methylated RNA. This synergy then accelerates the degradation of mRNA by directly interacting with YTHDF2 [46, 47].
eIF3
Generally, eukaryotic initiation factor 4 (eIF4) proteins, especially the cap-binding protein eIF4e, are required for translation initiation [48]. However, eIF3 was identified as a direct 5′UTR m6A-binding protein to initiate eIF4-independent translation [49]. About 35% of eIF3 Crosslinking and Immunoprecipitation (CLIP) sites mapped throughout the transcriptome overlap with m6A sites. Importantly, it is m6A, not m6Am that plays a part. Notably, METTL3 forms only m6A, not m6Am. Thus, 5′UTR m6A is probably an indicator of eIF3-dependent translation. A previous study showed m6A could directly bind and recruit eIF3 to the 5′UTR [49]. Recently, however, researchers have discovered a more complex and more ingenious mechanism for METTL3-mediated mRNA circulrization and passing eIF3 from 3′UTR to 5′UTR [44, 50]. Given m6A enrichment near 3′UTR [3], the latter is more convincing in the case of not ruling out the former.
HNRNPs
Heterogeneous nuclear ribonucleoprotein A2B1 (HNRNPA2B1), heterogeneous nuclear ribonucleoprotein C (HNRNPC), and heterogeneous nuclear ribonucleoprotein G (HNRNPG) are three abundant nuclear RNA-binding proteins responsible for hnRNA processing [51].
The previous understanding was that HNRNPA2B1 was identified to be a regulator in microRNA (miRNA) processing by selectively binding to the m6A site of miRNA directly [52]. Interestingly, recent studies concluded that HNRNPA2B1 depletion has minimal effect on mature miRNA levels in the central nervous system [53, 54], in contrast to earlier studies. These newer findings have challenged the idea that HNRNPA2B1 has a general role in microRNA processing. Santangelo et al. showed that HNRNPA2B1 selectively binds to GGAG or GGCU motifs on miRNA, suggesting that there is sequence-specific miRNA sorting into exosomes. This result is consistent with the uneven distribution of m6A modification in miRNA [55].
HNRNPC and HNRNPG also regulate mRNA splicing and abundance by processing m6A-modified RNA transcripts. The m6A site of hnRNA indirectly alters the binding of HNRNPC/G to its U-tract motifs, thereby modulating mRNA abundance and splicing [56, 57]. This phenomenon is termed ‘m6A-switch’.
Prrc2a
Wu and his colleagues discovered that Prrc2a is a novel m6A reader [58]. They found that Prrc2a deficiency in the nervous tissue leads to hypomyelination by affecting oligodendrocytes. Through transcriptome-wide analyses, they found that Prrc2a directly regulates Olig2 expression in an m6A-dependent manner in vitro and in vivo, which is essential in oligodendrocyte development. One interesting point is that Prrc2a, with YTHDF2, compete for RNA binding. Another is that FTO, an m6A demethylase, can trigger hypomyelination by downregulating Olig2 mRNA in an m6A-dependent manner [58]. These findings may build a dynamic m6A regulating hypomyelination model, which needs further studies.
IGF2BPs
IGF2BPs, including IGF2BP1/2/3, also serve as a distinct family of m6A readers. Different from YTH domain proteins, IGF2BP1/2/3 recognize the m6A consensus sequence through the K homology domains. IGF2BPs binding m6A enhances the stability and translation efficiency of their targeted mRNAs, like MYC mRNA, under normal and stress conditions [59].
Two approaches of effect of m6A on the transcriptome
The effect of m6A modification on the transcriptome has been continuously deepened with the research progress of the YTH-domain protein family and the discovery of other novel m6A readers. We summarize two regulatory approaches—direct approach and indirect approach—to systematically describe the possible regulatory mechanisms of m6A modification in the transcriptome.
Direct regulatory approach
Some m6A readers can directly recognize m6A modification on targeted transcripts and regulate the expression of corresponding targeted proteins (Fig. 3). The direct approach includes the regulation of mRNA translation efficiency and the regulation of mRNA abundance. On the one hand, through two possible mechanisms of eIF3 initiating translation and YTHDC2 loosening the spacial structure of mRNA, m6A-mRNA translation efficiency is improved. On the other hand, loosening mRNA is easier to be degraded. YTHDC1-mediated MAT2A mRNA degradation and YTHDF2 recruiting CCR4-NOT deadenylase complexes can reduce mRNA abundance.

Direct pathway of m6A to regulate transcriptome a Two possible mechanisms of eIF3 regulating mRNA translation initiation. ①: m6A directly promotes the recruitment of eIF3 and then induce translation initiation. ②: m6A delivers eIF3 to the 5′UTR through METTL3–eIF3h mediated mRNA circularization and induce translation initiation. b Approach of YTH-domain proteins(except YTHDC1) regulating mRNA stability and metabolism. ③: YTHDC1 reads MAT2A m6A, which induces SAM-mediated MAT2A mRNA degradation. ④ → ⑤/⑥: YTHDC2 binds m6A and unpacks the target m6A-mRNA to enhance the translation efficiency and degradation. ⑦: YTHDF1 binds to the m6A site surrounding the stop codon, then cooperates with the translation initiation machinery to enhance the translation efficiency of target RNAs. ⑧: YTHDF2 accelerates the degradation of m6A-modified RNA by directly recruiting CCR4-NOT deadenylase complexes. ⑨⑩: YTHDF3 can accelerate the degradation of mRNA by directly interacting with YTHDF2 while Prcc2a can decelerate this degradation. ⑪: YTHDF3 can synergize with YTHDF1 to promote the translation of m6A-RNA
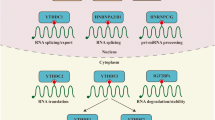
Indirect regulatory approach
Instead of binding m6A modification on mRNA directly, the expressions of some proteins can be modulated by upstream events, such as splicing, or upstream regulators like non-coding RNA (Fig. 4). The indirect pathway mainly occurs in the nucleus and plays an important role in mRNA selective splicing, non-coding RNA maturation, and RNA nuclear export. HNRNPC and HNRNPG form ‘m6A switch’ to alter the splicing sites in U-tract motifs. HNRNPA2B1 promotes miRNA maturation. YTHDC1 regulates RNA splicing through influencing recruitment of different splicing factors and facilitates RNA nuclear export.

Approach of m6A readers regulating on transcriptome indirectly. ①/①’: ‘m6A-switch’: HNRNPC and HNRNPG binding to the m6A site of hnRNA indirectly alters the connection of HNRNPC/G to its U-tract motifs, thereby modulating mRNA abundance and splicing. ②: HNRNPA2B1 selectively binds to the m6A site of pre-microRNAs and modulates the matureness of microRNA. ③: YTHDC1 promotes exon inclusion by recruiting or restricting different splicing factors, such as SRSFs. ④: YTHDC1 promotes the nuclear export of m6A-methylated mRNAs
Opposite roles of m6A in tumorigenesis
As we know, the effect of m6A on transcription is duplex, and through many different mechanisms, some of which are currently unknown to us. The dynamic regulation of m6A and the dynamic effects of m6A regulators determine that the overall impact of m6A on cells and tissues is specific, and m6A’s regulation of targeted protein expression also changes dynamically with changes in cell development stages.
We have compiled many studies on the effects of m6A writer, eraser, and reader on human tumor cells in recent years. We can see that the same m6A regulator can play an oncogenic role in some cancers, whereas a suppressing role in other cancers. (Table 1). Increasingly evidence showed that the levels and effects of m6A in tumor cells change continuously as the tumor progresses. For instance, changes in cellular m6A levels can alter the phenotypes of breast cancer cells [60] and the malignancy of gastric cancer [61].
Given the complexity of m6A’s regulation of the transcriptome, this is understandable and worth investigating its cause deeply. The contradictory results of some studies remind us that we cannot simply attribute the effects of m6A to certain tumors as oncogenic or suppressive. Therefore, we must carefully consider and pay attention to the overall impact and dynamic effects when introducing m6A modification to human cancer treatment.
Conclusion and future prospectus
Although current research on m6A modification has not had a massive impact on the practice of cancer diagnosis and treatment, great efforts have continued.
The first hotspot is m6A-associated cancer phenotyping, staging, and prognosis. A large number of small sample studies are widely involved in various human cancers, including lung cancer [72, 73], gastric cancer [61, 74], breast cancer [60, 68], glioma [75], urogenital cancer [76] etc.
Secondly, research on anticancer treatment related to m6A is also progressing. The intervention of m6A regulators of tumor cells can become a new target for adjuvant radiotherapy and chemotherapy. Research demonstrated that the upregulation of METTL14 expression in pancreatic cancer cells could reduce the response to mTOR signal-mediated autophagy after cisplatin treatment [77]. Other studies showed that m6A writer METTL3 could promote chemo-/radio-resistance in pancreatic cancer cells [78] and glioma stem-like cells [79]. More gratifyingly, scientists have been researching drugs associated with the regulation of m6A modification. One example is the ethyl ester form of Meclofenamic acid (MA), MA2—a highly selective inhibitor of FTO [80]. MA2 suppresses glioblastoma progression and prolongs lifespan in vivo [81], which suggests that m6A methylation could be a promising target for anti-glioblastoma therapy.
Tumor mutation burden (TMB), microsatellite instability (MSI), and Epstein‐Barr virus (EBV) were recognized as markers for cancer immunotherapy. A research conducted by Zhang et al. suggested that m6A modification was positively correlated with TMB/MSI status, and might be involved in immune responses of gastric cancer [61]. EBV-associated tumorigenesis has also been confirmed to require epitranscriptome reprogramming by METTL14 [82]. Thereby exploration of novel immunotherapy strategies targeting m6A is worth in-depth.
Future research on m6A RNA still faces many challenges
The first debate requiring to be settled is that whether those proposed functions of m6A regulators are dependent on m6A modification. Lin and colleagues identified that both wild-type and catalytically inactive METTL3 can boost the translation of some particular mRNAs such as epidermal growth factor receptor (EGFR) and the Hippo pathway effector TAZ, thus promoting the development of lung cancer [83]. Therefore, the direct causal relationship between m6A and cancer progression remains to be further established.
Another problem that needs to be solved is to clarify the selection preference of m6A. That means not only will we need to clarify the site selection mechanism of m6A modification, but also the selective binding mechanism among different m6A readers to m6A. Some new studies demonstrated that YTHDFs proteins exhibit virtually identical RNA-binding preferences at m6A sites [37]. Therefore, unknown mechanisms or new regulators that determine the selective regulation on transcriptome by m6A.
There is no doubt that much work lies ahead to acquire a complete picture of m6A epigenetics and understand how to make it benefit humanity practically.
Availability of data and materials
Not applicable.
References
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. P Natl Acad Sci USA. 1974;71(10):3971–5.
Dan D, Sharon M, Schraga S, Mali S, Lior U, Sivan O, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Nian L, Tao P. Probing RNA modification status at single-nucleotide resolution in total RNA. Method Enzymol. 2015;560:149–59.
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–72.
Narayan P, Ludwiczak RL, Goodwin EC, Rottman FM. Context effects on N6-adenosine methylation sites in prolactin mRNA. Nucleic Acids Res. 1994;22(3):419–26.
Shukun L, Liang T. Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. P Natl Acad Sci USA. 2014;111(38):13834–9.
Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem. 1994;269(26):17697–704.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3(11):1233–47.
Liao S, Sun H, Xu C. YTH domain: a family of N6-methyladenosine (m6A) readers. Genomics Proteomics Bioinform. 2018;16(02):99–107.
Guifang J, Ye F, Xu Z, Qing D, Guanqun Z, Ying Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang C, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Li J, Han Y, Zhang H, Qian Z, Jia W, Gao Y, et al. The m6A demethylase FTO promotes the growth of lung cancer cells by regulating the m6A level of USP7 mRNA. Biochem Bioph Res Co. 2019;512(3):479–85.
Zhuang C, Zhuang C, Luo X, Huang X, Yao L, Li J, et al. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO—PGC -1α signalling axis. J Cell Mol Med. 2019;23(3):2163–73.
Chen J, Sun Y, Xu X, Wang D, He J, Zhou H, et al. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16(23):2259–71.
Matthieu S. Structural chemistry of human RNA methyltransferases. ACS Chem Biol. 2016;11(3):575–82.
Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, et al. MTA is an arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20(5):1278–88.
Agarwala SD, Blitzblau HG, Hochwagen A, Fink GR. RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLoS Genet. 2012;8(6):e1002732.
Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Am Assoc Advanc Sci. 2015;347(6225):1002–6.
Narayan P, Rottman FM. An in vitro system for accurate methylation of internal adenosine residues in messenger RNA. Science. 1988;242(4882):1159–62.
Hengyou W, Huilin H, Huizhe W, Xi Q, Simen ZB, Lei D, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. 2018;22(2):191–205.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of mettl3 and mettl14 methyltransferases. Mol Cell. 2016;63(2):306–17.
Paweł Ś, Martin J. Structural insights into the molecular mechanism of the m(6)A writer complex. ELIFE. 2016;5:e18434.
Xiang W, Jing F, Yuan X, Zeyuan G, Delin Z, Zhu L, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534(7608):575–8.
Xiao-Li P, Bao-Fa S, Lu W, Wen X, Xin Y, Wen-Jia W, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P, et al. ALKBH5 inhibits pancreatic cancer motility by decreasing long non-coding RNA KCNK15-AS1 methylation. Cell Physiol Biochem. 2018;48(2):838–46.
Chongyuan W, Yuwei Z, Hongyu B, Yiyang J, Chao X, Jihui W, et al. A novel RNA-binding mode of the YTH domain reveals the mechanism for recognition of determinant of selective removal by Mmi1. Nucleic Acids Res. 2016;44(2):969–82.
Jianzhao L, Yanan Y, Dali H, Xiao W, Ye F, Liang Z, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Yuko U, Ikumi O, Yasuyuki F, Kaori K, Megumi K, Kentaro J, et al. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep-UK. 2017;7:42271.
Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, Gross SS. Reversible methylation of m 6 A m in the 5′ cap controls mRNA stability. Nature. 2017;541(7637):371–5.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N 6 -methyladenosine RNA demethylase. Cancer Cell. 2017;31(1):127–41.
Peter S, Ilona R, Stefan S. YTH: a new domain in nuclear proteins. Trends Biochem Sci. 2002;27(10):495–7.
Shukun L, Liang T. Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. P Natl Acad Sci USA. 2014;111(38):495–7.
Theler D, Dominguez C, Blatter M, Boudet J, Allain FH. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 2014;42(22):13911–9.
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10(11):927–9.
Xiao W, Adhikari S, Dahal U, Chen Y, Hao Y, Sun B, et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(6):507–19.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P, He E. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife. 2017;6:e31311.
Hiroki S, Mitsuyo M, Yuma I, Masayuki E, Akihiko M, Yuho S, et al. S-Adenosylmethionine synthesis is regulated by selective N6-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 2017;21(12):3354–63.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27(9):1115–27.
Bailey AS, Batista PJ, Gold RS, Grace Chen Y, de Rooij DG, Chang HY, Fuller MT (2017) The conserved RNA helicase YTHDC2 regulates the transition from proliferation to differentiation in the germline. Elife 6:e26116
Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS. Regulation of m6A transcripts by the 3ʹ→ 5ʹ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68(2):374–87.
Jain D, Puno MR, Meydan C, Lailler N, Mason CE, Lima CD, Anderson KV, Keeney S. ketu mutant mice uncover an essential meiotic function for the ancient RNA helicase YTHDC2. Elife. 2018;7:e30919.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N 6 -methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, Wu L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nature communications. 2016;7:12626.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N 6-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Li A, Chen Y, Ping X, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27(3):444–7.
Jackson RJ, Hellen CUT, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Bio. 2010;11(2):113–27.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m(6)A promotes cap-independent translation. Cell. 2015;163(4):999–1010.
Junho C, Shuibin L, Wencai Z, Qi L, Longfei W, Julia R, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561(7724):556–60.
Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135(8):851–67.
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–308.
Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482–5.
Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, et al. Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron. 2016;92(4):780–95.
Laura S, Giorgio G, Carla C, Claudia M, Carmine M, Roberta T, et al. The RNA-binding protein SYNCRIP is a component of the hepatocyte exosomal machinery controlling MicroRNA sorting. CELL REP. 2016;17(3):799–808.
Nian L, Qing D, Guanqun Z, Chuan H, Marc P, Tao P. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic acids research. 2017;45(10):6051–63.
Wu R, Li A, Sun B, Sun J, Zhang J, Zhang T, et al. A novel m 6 A reader Prrc2a controls oligodendroglial specification and myelination. Cell Res. 2019;29(1):23–41.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Fn J, Lb A, Io R, Ck R, Hc L, Mk D. N6-methyladenosine contributes to cellular phenotype in a genetically-defined model of breast cancer progression. Oncotarget. 2018;9(58):31231–43.
Zhang C, Zhang M, Ge S, Huang W, Lin X, Gao J, et al. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med-US. 2019;8(10):4766–81.
Ru D, Yikan C, Shubiao Y, Jianwei Z, Runqing H, Peisi L, et al. m6A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. Oncotargets Ther. 2019;12:4391–402.
Li T, Hu P, Zuo Z, Lin J, Li X, Wu Q, et al. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. BioMed Central. 2019;18(1):1–15.
Wen P, Jie L, Ranran C, Qiou G, Peng Y, Wenwei Q, et al. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res. 2019;38(1):393.
Xiao L, Jingyuan T, Wen H, Feng W, Pu L, Chao Q, et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8(56):96103–16.
Xiaoli C, Xiao W, Can C, Yuen G, Shuqin Z, Zhe Y, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7 g. Cancer Lett. 2018;415:11–9.
Hong W, Bei X, Jun S. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722:144076.
Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BioMed Central. 2019;19(1):326.
Zhe Y, Jiong L, Guoxing F, Shan G, Yuan W, Shuqin Z, et al. MicroRNA-145 modulates N 6-Methyladenosine levels by targeting the 3′-untranslated mRNA region of the N 6-methyladenosine binding YTH domain family 2 protein. J Biol Chem. 2017;292(9):3614–23.
Chen M, Wei L, Law CT, Tsang FHC, Shen J, Cheng CLH, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70.
Li Z, Dan L, Meifang Z, Cuiling Z, Xinchun L, Ruhua Z, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–61.
Jin G, Zhu M, Yin R, Shen W, Liu J, Sun J, et al. Low-frequency coding variants at 6p21.33 and 20q11.21 are associated with lung cancer risk in Chinese populations. Am J Human Gene. 2015;96(5):832–40.
Shi R, Yu X, Wang Y, Sun J, Sun Q, Xia W, et al. Expression profile, clinical significance, and biological function of insulin-like growth factor 2 messenger RNA-binding proteins in non–small cell lung cancer. Tumor Biol. 2017;39(4):115–32.
Li Y, Zheng D, Wang F, Xu Y, Yu H, Zhang H. Expression of demethylase genes, FTO and ALKBH1, is associated with prognosis of gastric cancer. Digest Dis Sci. 2019;64(6):1503–13.
Xi Z, Xue Y, Zheng J, Liu X, Ma J, Liu Y. WTAP expression predicts poor prognosis in malignant glioma patients. J Mol Neurosci. 2016;60(2):131–6.
João L, Daniela B, Rui H, Carmen J. The emerging role of epitranscriptomics in cancer: focus on Urological tumors. Genes-Basel. 2018;9(11):552.
Kong F, Liu X, Zhou Y, Hou X, He J, Li Q, et al. Downregulation of METTL14 increases apoptosis and autophagy induced by cisplatin in pancreatic cancer cells. Int J Biochem Cell Biol. 2020;122:105731.
Kosuke T, Masamitsu K, Ayumu A, Jun K, Masayasu T, Taroh S, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52(2):621–9.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m6A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37(4):522–33.
Yue H, Jingli Y, Qi L, Jiafei L, Shouzhe G, Hu Z, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43(1):373–84.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–34.
Lang F, Singh RK, Pei Y, Zhang S, Sun K, Robertson ES. EBV epitranscriptome reprogramming by METTL14 is critical for viral-associated tumorigenesis. PLoS Pathog. 2019;15(6):e1007796.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. P Natl Acad Sci USA. 2016;113(14):2047–56.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (81502525); the Science and Technology Development Project in Jinan (201907018).
Author information
Authors and Affiliations
Contributions
NZ designed and funded the study. GY, ZS were involved in discussion. GY, ZS drafted the manuscript and revised the manuscript. NZ critically analyzed and corrected the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, G., Sun, Z. & Zhang, N. Reshaping the role of m6A modification in cancer transcriptome: a review. Cancer Cell Int 20, 353 (2020). https://doi.org/10.1186/s12935-020-01445-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01445-y