
Abstract
Background
Endometrial cancer was the commonest gynecological malignancy in developed countries. Despite striking advances in multimodality management, however, for patients in advanced stage, targeted therapy still remained a challenge. Our study aimed to investigate new biomarkers for endometrial cancer and establish a novel risk score system of immune genes in endometrial cancer.
Methods
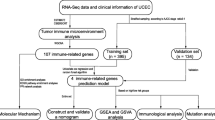
The clinicopathological characteristics and gene expression data were downloaded from The Cancer Genome Atlas (TCGA) database. Differentially expressed genes (DEGs) of immune genes between tumors and normal tissues were identified. Protein–protein interaction (PPI) network of immune genes and transcriptional factors was integrated and visualized in Cytoscape. Univariate and multivariate analysis were employed for key genes to establish a new risk score system. Receiver operating characteristic (ROC) curve and survival analysis were performed to investigate the prognostic value of the model. Association between clinical characteristics and the model was analyzed by logistic regression. For validation, we identified 34 patients with endometrial cancer from Fudan University Shanghai Cancer Center (FUSCC). We detected 14-genes mRNA expression and calculated the risk scores of each patients and we performed survival analysis between the high-risk group and the low-risk group.
Results
23 normal tissues and 552 tumor tissues were obtained from TCGA database. 410 immune-related DEGs was identified by difference analysis and correlation analysis. KEGG and GO analysis revealed these DEGs were enriched in cell adhesion, chemotaxis, MAPK pathways and PI3K-Akt signaling pathway, which might regulate tumor progression and migration. All genes were screened for risk model construction and 14 hub immune-related genes (HTR3E, CBLC, TNF, PSMC4, TRAV30, PDIA3, FGF8, PDGFRA, ESRRA, SBDS, CRHR1, LTA, NR2F1, TNFRSF18) were prognostic in endometrial cancer. The area under the curve (AUC) was 0.787 and the high-risk group estimated by the model possessed worse outcome (P < 0.001). Multivariate analysis suggested that the model was indeed an independent prognostic factor (high-risk vs. low-risk, HR = 1.14, P < 0.001). Meanwhile, the high-risk group was prone to have higher grade (P = 0.002) and advanced clinical stage (P = 0.018). In FUSCC validation set, the high-risk group had worse survival than the low-risk group (P < 0.001).
Conclusions
In conclusion, the novel risk model of immune genes had some merits in predicting the prognosis of endometrial cancer and had strong correlation with clinical outcomes. Furthermore, it might provide new biomarkers for targeted therapy in endometrial cancer.
Similar content being viewed by others


Background
Endometrial cancer was the leading gynecological malignancy in western countries and ranked sixth in women cancer worldwide [1, 2]. There were estimated 63,230 new cases and 11,350 deaths in female uterine corpus carcinoma in 2018 [3] and the incidence was still rising [4, 5]. Most newly diagnosed patients had favorable prognosis due to the early stage with 5-year survival rate over 80% [6]. However, patients with delayed diagnosis were proved to have dismal survival [7].
Uteri corpus endometrial carcinoma (UCEC) was often classified into two histological type: type I (estrogen-dependent endometrioid adenocarcinomas) and type II (estrogen-independent serous carcinomas). Type I carcinomas represented 80% of endometrial cancer with favorable prognosis and good response to estrogen treatment. Conversely, type II tumors, insensitive to estrogen, only accounting for 10–20%, had extremely aggressive behavior as advanced stage, distant metastasis [8].
Multimodality strategies, surgery followed by adjuvant therapy, had achieved great success in early-staged patients. Nevertheless, the treatment and management of advanced stage and recurrent patients still remained a challenge. Pre-clinical and clinical investigations of targeted therapies suggested efficacy for some agents. Single agent targeted therapies, however, had modest activity. Identifying potential biomarkers that effectively responded to targeted therapy appeared extremely urgent.
There were rarely specific gene alterations investigated in endometrial cancer. Mismatch repair genes (typically MLH1, MSH2, PMS2, or MSH6) mutation had been reported in Lynch syndrome related endometrial cancer [7]. BAF250a expression, also known as ARID1A, was commonly detected absent in high-grade endometrioid endometrial cancer at about 40% [9]. CTNNB1, commonly mutated in low grade, early stage endometrial cancer, were associated with worse recurrence-free survival [10]. FGFR2 mutation was also reported mutated in endometrial cancer [11]. Human epidermal growth factor receptor 2 (HER2)/neu, a receptor was discovered overexpressed in 30% of uterine serous carcinoma [12]. P53 protein, if absent or diffusely overexpressed, was associated with poor prognosis [8].
Recently, TCGA research group performed an integrating genomic characterization of endometrial cancer by whole-exome sequence analysis and proposed to divide it into four subgroups as “POLE ultra-mutated”, “hypermutated/microsatellite unstable”, “copy number”, “low/microsatellite stable” and “copy number high (serous-like)” [13,14,15]. Above this, in our study, we were devoted to exploring new biomarkers and establishing a risk score model to predict prognosis, aiming to provide novel therapeutic options of personalized medicine for endometrial cancer.
Methods
Data collection and enrichment analysis
Total 23 normal cases and 552 tumor cases with clinicopathological characteristics and expression data were downloaded from TCGA official website for the Uterine Corpus Endometrial Carcinoma projects (UCEC). 2498 immune-related genes data was downloaded from IMMPORT website. 410 immune genes were verified with |Log Fc| ≥ 2 and FDR < 0.25 by differential analysis. Gene Ontology (GO) analysis of these DEGs between UCEC and normal tissues was performed using blast2GO with P-value ≤ 1 and pathway enrichment analysis was carried out against the KEGG database with Q-value ≤ 1.
PPI network and survival analysis
The data of transcriptional factors (TFs) was obtained from Cistrome database. Differential expressed TFs were identified by the criteria of |Log FC| ≥ 1 and P value < 0.05. PPIs with a confidence score ≥ 0.4 and P value < 0.001 were reserved and further visualized in Cytoscape.
For survival analysis, we deleted partial cases with incomplete follow-up information, finally, 544 patients were reserved for further analysis. Univariate analysis was conducted to identify candidate genes which were significantly correlated with survival with P value < 0.05. Multivariate Cox hazards regression model was established to select independent prognostic genes. We constructed a prognostic gene signature according to a linear combination of gene expression values multiplied by a regression coefficient (β) accessed from the multivariate Cox proportional hazards regression model of each gene. The formula is as follows: risk score = expression of gene1 × β1gene1 + expression of gene2 × β2gene2 +… expression of genen × βngenen [16, 17]. All patients were divided into low- or high-risk groups according to the median risk score. Receiver operating characteristic (ROC) curve analysis by R package was performed to assess the predictive accuracy of the prognostic value for time-dependent cancer death.
The area under the curve (AUC) was calculated to measure the predictive ability of the gene signature for clinical outcomes [18].
Validation of the model
We obtained 34 patients diagnosed with endometrial cancer from Fudan University Shanghai Cancer Center (FUSCC) between January 2017 and February 2018. RNAs were distracted from total 34 tumor samples and cDNAs of these samples were synthesized by reverse transcription reaction kit (Takara, RR036A). Then we performed real time polymerase chain reaction (RT-PCR) (Takara, RR820A) to estimate 14 genes mRNA expression level in endometrial cancer. The internal reference was GAPDH mRNA. The primer sequences for 14 genes mRNA and GAPDH mRNA were listed in Additional file 1: Table S1. We calculated risk scores of each patients according to the 14 genes mRNA expression level observed to the formula of the model, and then we divided all 34 patients into the two groups: the high-risk group and the low-risk group according to the median value of risk scores. Survival analysis was performed in the two groups.
Statistical analysis
All statistical analysis were conducted by software R (v.3.6.1). The association between clinicopathologic characteristics and risk score system was analyzed by Wilcoxon signed-rank test and logistic regression. Kaplan–Meier method and Cox multivariate model were used for survival analysis. The cut-off value of risk score was determined by the median value.
Results
Identification of immune-related DEGs and functional annotation
Gene expression data was downloaded from TCGA database. 23 normal tissues and 552 tumor tissues were obtained and 6268 DEGs by difference analysis were shown in heatmap and volcano plot (in Fig. 1a, b). To identify immune-related genes of DEGs, 2498 immune genes data was downloaded from IMMPORT website. Eventually, 410 immune-related DEGs were obtained (shown in Fig. 1c, d, in Additional file 1: Table S2).

Identification of DEGs. a, b Heatmap and volcano plots of 6268 DEGs in endometrial cancer and normal tissues from TCGA database. c, d Heatmap and volcano plots of 410 immune-related DEGs. The colors in the heatmaps from green to red represent expression level from low to high. The red dots in the volcano plots represent up-regulation, the green dots represent down-regulation and black dots represent genes without differential expression
Moreover, we performed KEGG and GO enrichment analysis to elucidate the functional role of DEGs. The results of GO analysis revealed that “positive regulation of cell adhesion”, “positive regulation of protein kinase B signaling”, and “positive regulation of chemotaxis”, “regulation of leukocyte migration” were significantly enriched biological processes and pathways, which might be associated with tumor migration and tumor microenvironment (in Fig. 2a, b). KEGG enrichment analysis manifested that these genes were probably enriched in cytokine–cytokine receptor interaction, PI3K-Akt signaling pathway, MAPK signaling pathway and Ras signaling pathway which might regulate cancer progression (in Fig. 2c, d).

GO and KEGG enrichment analysis of DEGs. a, b GO analysis. GO analysis divided DEGs into three functional groups: molecular function (MF), biological processes (BP), and cell composition (CC). c, d KEGG analysis of DEGs
Interaction network for immune-specific genes and transcription factors
To find out the transcriptional factors (TFs) for immune-related genes, information on TFs were downloaded from Cistrome database. We investigated differentially expressed TFs with Log FC ≥ 2 and FDR < 0.25 (shown in Fig. 3a, b) and selected TFs relative to immune genes by correlation analysis with the filter for correlation coefficient = 0.04 and P value = 0.001 (data was shown in Additional file 2: Table S2). Integration of protein–protein interaction (PPI) networks was visualized in software Cytoscape (in Fig. 3c).

Interaction network for immune-specific genes and transcription factors. a, b Heat map and Volcano plots of differentially expressed transcription factors (TFs). The colors in the heatmaps from green to red represent expression level from low to high. The red dots in the volcano plots represent up-regulation, the green dots represent down-regulation and black dots represent TFs without differential expression. c A significant module from protein–protein interaction network
Clinical characteristics in TCGA database and FUSCC
Baseline characteristics were downloaded from TCGA database. Patients with incomplete follow-up information were deleted and total 544 patients were identified. In this cohort, shown in Table 1, the median overall survival time was 28 months (1–229 months). Age at diagnosis ranged from 31 to 90 years with median was 64 years. Majority of patients (n = 407, 74.82%) were endometrioid endometrial adenocarcinoma, followed by serous endometrial adenocarcinoma (n = 115, 21.14%) and mixed type was least (n = 22, 4.04%). 40.63% were low/moderate and 59.38% were high grade. 428 (84.42%) patients were founded tumor free but 79 (15.58%) were with tumor. As for lymph node, only 16.74% (n = 74) possessed positive pelvic lymph node and 10.30% were founded positive para-aortic lymph node. Stage I accounted for 62.32% (n = 339), stage II was 9.56% (n = 52) and stage III was 22.61% (n = 123) and stage IV was 5.51% (n = 30). For validation set, we obtained 34 patients with endometrial cancer from FUSCC. Clinicopathological characteristics of these patients were shown in Additional file 3: Table S3.
Construction of risk score system
Univariate and multivariate survival analysis by Cox proportional hazards models were conducted to select prognostic key genes. All the genes with significant P values were screened for risk model construction. And the risk score algorithm comprised of 14 hub genes (HTR3E, CBLC, TNF, PSMC4, TRAV30, PDIA3, FGF8, PDGFRA, ESRRA, SBDS, CRHR1, LTA, NR2F1, TNFRSF18) was established in Table 2. Otherwise, other 8 genes in this model including VIPR2, LGR5, IL13RA2, PTN, BACH2, GHR, ADCYAP1R1, ORM1 were not significant with overall survival. Risk scores could be calculated as: HTR3E * 0.513 + CBLC * 0.015 + TNF * 0.034 + PSMC4 * 0.004 + TRAV30 * 0.110 − PDIA3 * 0.004 + FGF8 * 0.020 + PDGFRA * 0.029 + ESRRA * 0.048 + SBDS * 0.016 + CRHR1 * 0.162− LTA * 0.819 + NR2F1 * 0.019 − TNFRSF18 * 0.028. In this gene model, three of them (PDIA3, LTA, TNFRSF18) were negatively correlated with survival and other 11 genes were positively related to overall survival. The contribution of each gene made to this risk score model was weighted by the value of coefficients. The risk score for each patient was estimated according to the expressions of the 14 hub genes (shown in Fig. 4a).

Construction of risk score system. a Risk score estimated of each patient on the basis of 14 hub genes expression. b ROC curve for the risk model. c Patients were divided into high risk group and low risk group. Survival analysis of the patients is also shown
All patients scored by the model could be classified into two groups: the high-risk group and low-risk group (in Fig. 4c). Receiver operating characteristics (ROC) curve was constructed to assess the predictive accuracy and the area under the curve (AUC) was 0.787 (in Fig. 4b). Similarly, results of survival analysis revealed that the high-risk group was associated with worse outcome than the low-risk group (P < 0.001).
Prognostic value of the risk model and clinical association
To validate the prognostic value of this risk model, survival analysis was performed by univariate and multi-cox hazards regression model (in Fig. 5a, b). In Table 3, we discovered that age at diagnosis (≥ 34 year vs. < 34 years, HR = 1.06, P = 0.006), tumor status (with tumor vs tumor free, HR = 5.50, P < 0.001), peritoneal cytology (positive vs. negative, HR = 4.62, P = 0.003), pelvic lymph node (positive vs. negative, HR = 4.20, P = 0.013) and risk model (high-risk vs. low-risk, HR = 1.14, P < 0.001) were independent prognostic factors by multivariate analysis.

Validation of the prognostic value of the risk model. a Univariate regression model. b Multi-cox hazards regression model
Furthermore, we also analyzed the association between risk model and clinical characteristics by logistic regression, and figured out that the high-risk group was prone to possess higher grade (P = 0.002) and advanced clinical stage (P = 0.018) in Fig. 6. Meanwhile, we investigated the correlation between each gene in this model and clinicopathological features separately (in Additional file 4: Figure S1).

Association between the risk model and different clinical characteristics
External validation of the model in FUSCC
We detected 14-genes mRNAs expression of 34 patients with endometrial cancer in FUSCC and calculated risk scores of each patients according to the formula of the model. All patients were classified into two groups: the high-risk group and the low-risk group by the median value of risk scores. We investigated that the high-risk group had worse survival than the low-risk group significantly with P < 0.001 in Fig. 7.

Survival analysis of the high-risk group and the low-risk group scored by the model in FUSCC validation set. All 34 patients were divided into two groups by the median value of risk scores: the high-risk group and the low-risk group
Discussion
In our study, we identified and constructed a 14 hub genes risk score model for endometrial cancer. All gene expression data and patients clinical characteristics information were downloaded from TCGA dataset. We analyzed the 6268 DEGs between endometrial cancer and normal tissues and integrated the immune genes from IMMPORT database, eventually, we verified 410 immune-related DEGs. Moreover, univariate and multi-cox regression were employed for the key genes, subsequently, a 14 hub-genes model including HTR3E, CBLC, TNF, PSMC4, TRAV30, PDIA3, FGF8, PDGFRA, ESRRA, SBDS, CRHR1, LTA, NR2F1, TNFRSF18, was successfully established. Furthermore, to investigate the prognostic value of the model, we performed the ROC curve and investigate the association between the model and clinical features. As expected, the high-risk group was correlated with worse overall survival and was inclined to have advanced stage and higher histological grade which might manifest poor outcome.
Several genes in our model had been studied in human cancers. CBLC (CBL proto-oncogene c), a homologue of CBL protein family, which could activate receptor tyrosine kinases (RTK), was frequently elevated in non-small cell lung cancer (NSCLC), but the function of CBLC in tumorigenesis remained still unknown [19]. PSMC4 (proteasome 26S subunit, ATPase, 4), was recognized as a house keeper gene in breast cancer [20], but was detected upregulated in prostate carcinoma, which might promote tumorigenesis [21]. Ye et al. [22] discovered that decreased PDIA3 (protein disulfide isomerase family A, member 3, also known as ERp57) expression could enhanced apoptosis, and suppressed proliferation, invasion, and migration of acute myeloid leukemia cells. Fibroblast growth factor 8 (FGF8) was recognized as an oncogene, and elevated gene expression in hormonal cancers such as prostate cancer and breast cancer, was associated with a poor prognosis [23]. Chang et al. [24] sequenced 10 samples and found out that oncogene PDGFRA (platelet-derived growth factor receptor, alpha polypeptide) was mutated in endometrial cancer. Similarly, mutations in PDGFRA and/or KIT were found in 5 endometrial carcinosarcomas (5/34, 14.7%) [25]. ESRRA (estrogen-related receptor alpha), which shared structural similarities with estrogen receptors, was reported to play a role in endometrial cancer tumorigenesis by Yoriki et al. [26]. Recently, they discovered that ESRRA could be a target of TGF-β to promote epithelial–mesenchymal transition in endometrial cancer. Positive correlations between Corticotropin releasing hormone receptor 1 (CRHR-1) and PR expression might be associated with more advanced FIGO stage disease in Miceli et al’ s findings [27]. Otherwise, Graziani et al. [28] revealed that CRH induced time- and concentration-dependent inhibition of Ishikawa cell growth through the cAMP-PKA pathway. Niwa et al. [29] detected Lymphotoxin-alpha (LTA) C804A and A252G polymorphisms in 110 endometrial cancer patients. They found out either one or two of the variant alleles was associated with a significantly lower risk of endometrial cancer (OR = 0.54, 95% CI 0.33–0.87, P = 0.012). Of the 14 genes, five genes as Serotonin (5-HT) receptors 3E subunits (HTR3E), TRAV30, SBDS, NR2F1 and TNFRSF18 were firstly reported in endometrial cancer.
Similar to our research, Liu et al. [30] constructed a panel of 7 DEG signatures consisting of PHLDA2, GGH, ESPL1, FAM184A, KIAA1644, ESPL1, and TRPM4 to predict the prognosis of endometrial cancer. Wang et al. [31] identified a six-gene model consisting of CTSW, PCSK4, LRRC8D, TNFRSF18, IHH, and CDKN2A by using robust likelihood‐based survival modeling for endometrial cancer. Besides, a lncRNA signature comprising LINC00491, LINC00483, ADARB2-AS1, and C8orf49 showed remarkable prognostic value in endometrial cancer [32].
Unlike previous researches, we firstly focused on the differentially expressed immune-related genes and set up and validated a novel immune-related signature for prognostic model. Tumor microenvironment in endometrial cancer had been elucidated by Sahoo et al. [33]. The dynamic interaction between tumor cells and microenvironment was essential for tumor proliferation, progression and migration. For instance, stromal estrogen receptor (ERα) mediated the mitogenic effects of estrogen on endometrial cell proliferation [34], the same to the gene ESRRA in this model. In our findings, we figured out several immune genes that might be correlated to tumor microenvironment and could provide guidance for the research on tumor microenvironment and tumor pathogenesis.
However, there were some limitations in our research. Firstly, the sample size in our study was small and a larger cohort and more abundant sequencing results were needed. Secondly, we only focused on the gene expression level, but ignored other events such as the gene mutation, methylation, and copy number amplification, which were also important in tumor progression.
Conclusions
In conclusion, we constructed and validated the prognostic value of a new risk score system, which might serve as a potential predictor for endometrial cancer. Our findings revealed that the model was strongly correlated with clinical characteristics and we guessed that high-risk patients might possess advanced stage and low/moderate grade. Furthermore, our data might have potential to guide personalized treatment and explore new biomarkers for targeted therapy in endometrial cancer.
Availability of data and materials
The data was available in The Cancer Genome Atlas database (https://cancergenome.nih.gov/); IMMPORT database (https://www.immport.org/) and the Cistrome Project (https://www.cistrome.org/).
Abbreviations
- UCEC:
-
Uteri corpus endometrial carcinoma
- TCGA:
-
The Cancer Genome Atlas
- DEGs:
-
Differentially expressed genes
- TF:
-
Transcriptional factors
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GO:
-
Gene Ontology
- PPI:
-
Protein–protein interaction
- ROC:
-
Receiver operating characteristic
- AUC:
-
The area under the curve
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30.
Njoku K, Chiasserini D, Whetton AD, Crosbie EJ. Proteomic biomarkers for the detection of endometrial cancer. Cancers. 2019;11(10):1572.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Morice P, Leary A, Creutzberg C, Abu-Rustum N, Darai E. Endometrial cancer. Lancet. 2016;387(10023):1094–108.
Amant F, Mirza MR, Koskas M, Creutzberg CL. Cancer of the corpus uteri. Int J Gynaecol Obstet. 2018;143(Suppl 2):37–50.
Urick ME, Bell DW. Clinical actionability of molecular targets in endometrial cancer. Nat Rev Cancer. 2019;19(9):510–21.
ACOG S. Practice Bulletin No. 149: endometrial cancer. Obstet Gynecol. 2015;125(4):1006–26.
Arend RC, Jones BA, Martinez A, Goodfellow P. Endometrial cancer: molecular markers and management of advanced stage disease. Gynecol Oncol. 2018;150(3):569–80.
Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW, Steidl C, Wiseman SM, Gascoyne RD, Gilks B, et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J Pathol. 2011;224(3):328–33.
Kurnit KC, Kim GN, Fellman BM, Urbauer DL, Mills GB, Zhang W, Broaddus RR. CTNNB1 (beta-catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod Pathol. 2017;30(7):1032–41.
Byron SA, Gartside M, Powell MA, Wellens CL, Gao F, Mutch DG, Goodfellow PJ, Pollock PM. FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS ONE. 2012;7(2):e30801.
Fader AN, Roque DM, Siegel E, Buza N, Hui P, Abdelghany O, Chambers SK, Secord AA, Havrilesky L, O’Malley DM, et al. Randomized phase II trial of carboplatin-paclitaxel versus carboplatin-paclitaxel-trastuzumab in uterine serous carcinomas that overexpress human epidermal growth factor receptor 2/neu. J Clin Oncol. 2018;36(20):2044–51.
Piulats JM, Matias-Guiu X. Immunotherapy in endometrial cancer: in the nick of time. Clin Cancer Res. 2016;22(23):5623–5.
Murali R, Delair DF, Bean SM, Abu-Rustum NR, Soslow RA. Evolving roles of histologic evaluation and molecular/genomic profiling in the management of endometrial cancer. J Natl Compr Cancer Netw. 2018;16(2):201–9.
Bell DW, Ellenson LH. Molecular genetics of endometrial carcinoma. Annu Rev Pathol. 2019;14:339–67.
Liu X, Wu J, Zhang D, Bing Z, Tian J, Ni M, Zhang X, Meng Z, Liu S. Identification of potential key genes associated with the pathogenesis and prognosis of gastric cancer based on integrated bioinformatics analysis. Front Genet. 2018;9:265.
Lin T, Gu J, Qu K, Zhang X, Ma X, Miao R, Xiang X, Fu Y, Niu W, She J, et al. A new risk score based on twelve hepatocellular carcinoma-specific gene expression can predict the patients’ prognosis. Aging. 2018;10(9):2480–97.
Julich-Haertel H, Urban SK, Krawczyk M, Willms A, Jankowski K, Patkowski W, Kruk B, Krasnodebski M, Ligocka J, Schwab R, et al. Cancer-associated circulating large extracellular vesicles in cholangiocarcinoma and hepatocellular carcinoma. J Hepatol. 2017;67(2):282–92.
Hong SY, Kao YR, Lee TC, Wu CW. Upregulation of E3 ubiquitin ligase CBLC enhances EGFR dysregulation and signaling in lung adenocarcinoma. Cancer Res. 2018;78(17):4984–96.
Szabo A, Perou CM, Karaca M, Perreard L, Palais R, Quackenbush JF, Bernard PS. Statistical modeling for selecting housekeeper genes. Genome Biol. 2004;5(8):R59.
Hellwinkel OJ, Asong LE, Rogmann JP, Sultmann H, Wagner C, Schlomm T, Eichelberg C. Transcription alterations of members of the ubiquitin-proteasome network in prostate carcinoma. Prostate Cancer Prostatic Dis. 2011;14(1):38–45.
Ye Q, Fu P, Dou J, Wang N. Downregulation of PDIA3 inhibits proliferation and invasion of human acute myeloid leukemia cells. Onco Targets Ther. 2018;11:2925–35.
Mattila MM, Harkonen PL. Role of fibroblast growth factor 8 in growth and progression of hormonal cancer. Cytokine Growth Factor Rev. 2007;18(3–4):257–66.
Chang YS, Huang HD, Yeh KT, Chang JG. Genetic alterations in endometrial cancer by targeted next-generation sequencing. Exp Mol Pathol. 2016;100(1):8–12.
Biscuola M, Van de Vijver K, Castilla MA, Romero-Perez L, Lopez-Garcia MA, Diaz-Martin J, Matias-Guiu X, Oliva E, Palacios Calvo J. Oncogene alterations in endometrial carcinosarcomas. Hum Pathol. 2013;44(5):852–9.
Yoriki K, Mori T, Kokabu T, Matsushima H, Umemura S, Tarumi Y, Kitawaki J. Estrogen-related receptor alpha induces epithelial–mesenchymal transition through cancer-stromal interactions in endometrial cancer. Sci Rep. 2019;9(1):6697.
Miceli F, Ranelletti FO, Martinelli E, Petrillo M, Scambia G, Navarra P, Ferrandina G. Expression and subcellular localization of CRH and its receptors in human endometrial cancer. Mol Cell Endocrinol. 2009;305(1–2):6–11.
Graziani G, Tentori L, Portarena I, Barbarino M, Tringali G, Pozzoli G, Navarra P. CRH inhibits cell growth of human endometrial adenocarcinoma cells via CRH-receptor 1-mediated activation of cAMP-PKA pathway. Endocrinology. 2002;143(3):807–13.
Niwa Y, Ito H, Matsuo K, Hirose K, Ito N, Mizuno M, Hamajima N, Tajima K, Nakanishi T. Lymphotoxin-alpha polymorphisms and the risk of endometrial cancer in Japanese subjects. Gynecol Oncol. 2007;104(3):586–90.
Liu J, Zhou S, Li S, Jiang Y, Wan Y, Ma X, Cheng W. Eleven genes associated with progression and prognosis of endometrial cancer (EC) identified by comprehensive bioinformatics analysis. Cancer Cell Int. 2019;19:136.
Wang Y, Ren F, Chen P, Liu S, Song Z, Ma X. Identification of a six-gene signature with prognostic value for patients with endometrial carcinoma. Cancer Med. 2018;7(11):5632–42.
Xia L, Wang Y, Meng Q, Su X, Shen J, Wang J, He H, Wen B, Zhang C, Xu M. Integrated bioinformatic analysis of a competing endogenous RNA network reveals a prognostic signature in endometrial cancer. Front Oncol. 2019;9:448.
Sahoo SS, Zhang XD, Hondermarck H, Tanwar PS. The emerging role of the microenvironment in endometrial cancer. Cancers. 2018;10(11):408.
Chung D, Gao F, Jegga AG, Das SK. Estrogen mediated epithelial proliferation in the uterus is directed by stromal Fgf10 and Bmp8a. Mol Cell Endocrinol. 2015;400:48–60.
Acknowledgements
This investigation was supported by Natural Science Foundation of Shanghai [No.17ZR1406000]. We would like to thank Dr. Tianjiao Li and Dr. Yajie Lei for providing us with experimental support.
Funding
This article was supported by Natural Science Foundation of Shanghai [No. 17ZR1406000]. The funding agency had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
XC and HZ contributed to the conception of the study. HZ, CZ and HL wrote the main part of the manuscript. LC helped revise the manuscript with constructive suggestion. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have agreed for this publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Primer sequences of 14-genes mRNA for reverse transcriptional PCR.
Additional file 2: Table S2.
Differentially expressed immune genes between endometrial cancer and normal tissues.
Additional file 3: Table S3.
Clinicopathological characteristics of 34 patients with endometrial cancer in FUSCC.
Additional file 4: Figure S1.
The association between clinical variables and each genes in this risk-score system.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, H., Zhang, C., Li, H. et al. A novel risk score system of immune genes associated with prognosis in endometrial cancer. Cancer Cell Int 20, 240 (2020). https://doi.org/10.1186/s12935-020-01317-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01317-5