
Abstract
Background
Microbial production of chemicals from renewable carbon sources enables a sustainable route to many bioproducts. Sugar streams, such as those derived from biomass pretreated with ionic liquids (IL), provide efficiently derived and cost-competitive starting materials. A limitation to this approach is that residual ILs in the pretreated sugar source can be inhibitory to microbial growth and impair expression of the desired biosynthetic pathway.
Results
We utilized laboratory evolution to select Escherichia coli strains capable of robust growth in the presence of the IL, 1-ethyl-3-methyl-imidizolium acetate ([EMIM]OAc). Whole genome sequencing of the evolved strain identified a point mutation in an essential gene, cydC, which confers tolerance to two different classes of ILs at concentrations that are otherwise growth inhibitory. This mutation, cydC-D86G, fully restores the specific production of the bio-jet fuel candidate d-limonene, as well as the biogasoline and platform chemical isopentenol, in growth medium containing ILs. Similar amino acids at this position in cydC, such as cydC-D86V, also confer tolerance to [EMIM]OAc. We show that this [EMIM]OAc tolerance phenotype of cydC-D86G strains is independent of its wild-type function in activating the cytochrome bd-I respiratory complex. Using shotgun proteomics, we characterized the underlying differential cellular responses altered in this mutant. While wild-type E. coli cannot produce detectable amounts of either product in the presence of ILs at levels expected to be residual in sugars from pretreated biomass, the engineered cydC-D86G strains produce over 200 mg/L d-limonene and 350 mg/L isopentenol, which are among the highest reported titers in the presence of [EMIM]OAc.
Conclusions
The optimized strains in this study produce high titers of two candidate biofuels and bioproducts under IL stress. Both sets of production strains surpass production titers from other IL tolerant mutants in the literature. Our application of laboratory evolution identified a gain of function mutation in an essential gene, which is unusual in comparison to other published IL tolerant mutants.

Similar content being viewed by others

Background
To enable the widespread adoption of next-generation biofuels, microbial hosts must be evaluated for productivity under cost-effective production regimes that can compete in the commodity market. Lignocellulosic plant biomass is a promising renewable carbon source, but the solubilization of sugars from this recalcitrant biomass is a technical challenge [1, 2]. One promising solution to this problem involves pretreating the plant biomass with an ionic liquid (IL), which allows the rapid extraction of sugars with greater ease when compared with conventional methods [3]. The resulting sugars from this pretreatment are then used in a microbial conversion process to produce the desired target compound. However, trace concentrations of many commonly used ILs from the lignocellulosic-derived carbon are inhibitory to cell growth [4,5,6], thus necessitating additional costly wash steps to fully purify this sugar source [7]. Moreover, unlike ethanol, the expression of next-generation biofuels from synthetic gene pathways requires additional optimization for function in the presence of ILs [8]. Optimization of both tolerance to ILs, as well as productivity under these growth constraints is required to fully exploit the value of biomass pretreated with ILs. One current approach is to develop ILs with low downstream impacts on the microbial conversion process [6, 9,10,11]. However, another useful strategy has also been to generate microbial hosts tolerant to a broad range of ILs, simplifying downstream microbial conversion of carbon sources to the target compounds.
The development of efficient microbial conversion in the presence of residual ILs remains a challenge in the field. Previous studies in this area have seen limited success in optimizing components of the process in a piecemeal fashion, as they achieve robust stress tolerance or high production, but not both. Saccharomyces cerevisiae has been used with IL pretreated biomass, but for the production of fermentation products such as ethanol [12]. With regards to advanced biofuels such as d-limonene and bisabolene [13], reports from the bacterial host, E. coli, have described more relevant advances. For example, Ruegg et al. used a gene cassette from an IL tolerant bacterium Enterobacter lignolyticus to auto-regulate expression of an imidazolium responsive efflux pump, which when expressed in Escherichia coli, improved host strain viability and allowed production of the biodiesel precursor bisabolene in the presence of the ionic liquid [14]. An adaptive laboratory evolution study to generate IL tolerant strains of E. coli has resulted in the discovery of several cellular transporters [15], which may prove to be useful in engineering production strains in future studies. Frederix et al. expressed IL tolerant cellulases in yet another IL tolerant E. coli background to use unsaccharified cellulose as a carbon source. The same strain, in turn, produced d-limonene (hereafter referred to as limonene), a bio-jet fuel, in the presence of ILs [16]. While successful relative to that in wild-type microbial strain backgrounds, the production of either bisabolene or limonene from Ruegg et al. or Frederix et al. was less than the maximum level produced by the same strain under optimal conditions. These results suggest that the IL inhibition of the biosynthetic pathways had not been fully addressed. Thus, it remains a challenge to develop a microbial strain tolerant to residual ILs used to pretreat biomass and still maintain robust bioconversion pathway productivity.
In this study, we used laboratory evolution to identify mutants that confer tolerance to the most commonly used model IL, 1-ethyl-3-methyl-imidizolium acetate (hereafter referred to as [EMIM]OAc). [EMIM]OAc has many favorable characteristics that facilitate the treatment and processing of biomass for sugar extraction by a variety of methods [17]. Whole genome re-sequencing implicated cydC, a cytochrome assembly factor and component of an ABC transporter complex [18] as the candidate gene for the observed tolerance. We successfully produced two candidate biofuels (limonene and isopentenol) in the presence of [EMIM]OAc and characterized global proteomic changes that occur under [EMIM]OAc stress.
Results
Laboratory evolution identifies cydC-D86G, a mutant that confers tolerance to [EMIM]OAc
We identified an improved host chassis for tolerance to [EMIM]OAc using laboratory evolution while maintaining production conditions. Mutant strains in a wild-type E. coli background that were capable of growing in 100 mM [EMIM]OAc were recovered after only 48 h of selection with exogenous [EMIM]OAc (Fig. 1a). This concentration of [EMIM]OAc was selected because it represents a typical concentration of trace IL after three washes with water, as opposed to the full eight water/ethanol wash regimen required to reduce residual IL levels below detection levels [19]. In order to validate that these candidate mutants were genetic mutations and not the result of adaptation, we used a dynamic cultivation regime [20] to alternate rounds of growth in the presence or absence of [EMIM]OAc (Fig. 1b, c). Five different isolates from two different rounds were recovered from this screen, and strikingly, three of them had similar robust growth profiles when compared side by side (Fig. 1c).

Adaptation and Laboratory Evolution of E. coli DH1 in presence of [EMIM]OAc. a Exponential phase E. coli cells were grown in the presence of 100 mM [EMIM]OAc at 30 °C in a microtiter dish until a change in optical density was detected (“Methods”). Red circles, growth of wild-type cells in the absence of [EMIM]OAc. Black squares, growth of wild-type cells in the presence of [EMIM]OAc. Standard deviation from three biological replicates is indicated as the shaded region surrounding the red circles or black squares. Optical density is plotted on a log10 scale. b Schematic, behavior of cells after gaining a heritable mutation (top panel) compared to adaptation (bottom panel). Cells initially grown in the absence of the ionic liquid stress (“EZ”) are back-diluted into medium containing the ionic liquid (“EZ + IL”). After a long lag phase, an increase in optical density is detected. Cells are subsequently back diluted into fresh media without the ionic liquid (“EZ”) and then retested for growth in the presence of the ionic liquid (“EZ + IL”). A heritable mutation would prevent the appearance of an extended lag phase after the first exposure to an ionic liquid. If E. coli adapts to the IL, cells would show a similarly long lag phase as with the first treatment with ionic liquid. c Cells exhibit a heritable response to [EMIM]OAc. All three independent isolates show similar tolerance to [EMIM]OAc. Error bars indicate standard deviation from three biological replicates
To identify potential putative mutations, cells from the liquid cultures were streaked to single colonies on solid agar plates, and a single colony from each plate was selected at random to confirm tolerance to the IL. We generated genomic libraries of all five mutants recovered from this study. Libraries were subjected to MiSeq Illumina deep sequencing. Basepairs that differed from the parent strain were called with CNVnator [21] (“Methods”). As these spontaneous mutants were derived from independent liquid cultures started from independent single colonies, we expected to find different mutants in different chromosomal loci that led to similar phenotypes. Remarkably, a single hitherto unknown mutation was identified in all five libraries. The mutation identified across all five of these isolates, but absent in the parental strain, was an aspartic acid to glycine mutation at residue 86 of cydC, a membrane protein thought to be involved in cytochrome bd-I assembly or amino acid efflux into the periplasm [22,23,24,25,26]. There were conflicting reports regarding cydC being an essential or dispensable gene. Without this knowledge, it could not be deduced directly whether cydC-D86G was a loss-of-function or gain-of-function mutation [27, 28]. In the two IL-tolerant, but slower growing mutant strains, we also detected unique premature stop codons (amber mutations) in the open reading frame of rcdA. This is consistent with a rcdA mutation identified in a previous study, which could ameliorate IL toxicity but did not fully restore the limonene production titer [16]. The amino acid change recovered in cydC was a strong candidate for the increased tolerance to the model IL, [EMIM]OAc, and belongs to a different functional category from IL tolerance genes discovered thus far. To assess the benefits of both improved tolerance and production, we focused our study on understanding this mutant.
We first examined the effect of this mutation on microbial growth. To confirm that it is the mutant cydC-D86G that conferred tolerance to [EMIM]OAc, we constructed plasmids encoding wild-type cydC or the mutant, cydC-D86G, under the control of the inducible trc promoter and transformed these plasmids into the parental strain. Since concentrations higher than 4 µM IPTG resulted in growth inhibition when overexpressing wild-type PTRC-cydC (Additional file 1: Figure S1), subsequent experiments relied on leaky gene expression without addition of the inducer. Upon treatment with [EMIM]OAc, the strain carrying an additional wild-type copy of PTRC-cydC failed to grow when treated with 75 mM [EMIM]OAc (Fig. 2a). In contrast, the strain carrying an additional copy of PTRC-cydC-D86G was viable in the presence of 75 mM [EMIM]OAc (Fig. 2a). In addition to conferring tolerance to [EMIM]OAc, we also observed that the original DH1 cydC-D86G strains were also somewhat tolerant to a structurally different IL, ethanolamine acetate ([EOA]OAc Additional file 1: Figure S2).

cydC Is An Essential Gene, and Expression of cydC-D86G is sufficient to confer tolerance to [EMIM]OAc. a E. coli DH1 cells harboring a plasmid-borne copy of either wild type or mutant cydC-D86G [pTE42/Ptrc-cydD-cydC or pTE43/Ptrc-cydD-cydC-D86G] were untreated or exposed to 75 mM [EMIM]OAc (“Methods”). The change in OD600 was measured every fifteen minutes for 40 h. b Evidence that cydC is essential by plasmid shuffle assay. After generating a strain with both plasmid pTE50 and ∆cydC, cells are grown without selection to allow for spontaneous plasmid loss. The rate of plasmid loss in both wild type and ∆cydC strains is compared by selecting for cells without the plasmid on sucrose plates. To allow for spontaneous plasmid loss, cells were grown under non-selective conditions for 24 h at 30 °C. Cells were serially diluted after growth in nonselective media, and plated onto solid LB agar media with or without 10% sucrose. Colonies that are viable in the presence of sucrose have lost the plasmid
To maximize the ease of deploying this IL-tolerant gene to other strain backgrounds, we mutagenized the cydC-D86 codon and screened for additional point mutants which were also tolerant to [EMIM]OAc. This time, however, we used the native cydC promoter. Increased expression of cydC under its native promoter from a multi-copy plasmid had no impact on cell growth, corroborating reports from the literature [24]. From this screen we identified that cydC-D86V was also tolerant to [EMIM]OAc. As we did not modify the background genomic copy of cydC in these cells, this implied that the plasmid-borne cydC-D86V still conferred tolerance to [EMIM]OAc, even when wild-type cydC was potentially expressed in the cell. These results indicate that the cydC-D86G mutant by itself is sufficient to confer tolerance to [EMIM]OAc as well as to a lesser extent [EOA]OAc, and that this activity is distinct from an increased expression of the wild-type cydC gene. Furthermore, we identified that similar point mutants such as cydC-D86V can also confer tolerance to [EMIM]OAc. This allows for rapid deployment of the cydC mutant versions with a simple plasmid transformation, without requiring laborious genomic allele replacement strategies.
We also confirmed that the single point mutation (D86G) in cydC specifically provides the IL tolerance. One mechanism for the new function is that cydC-D86G encodes a loss-of-function mutation and thus inactivated cydC is responsible for the observed phenotype. A loss-of-function mutation can only be expected in genes that are not essential, since essential genes cannot be deleted. However, reports in the literature do not allow a clear assignment of this aspect for cydC. Gene deletions of cydC are absent from E. coli deletion collections [27, 29] suggesting that it is an essential gene required for E. coli viability. As such, a direct gene-deletion approach using a selectable drug cassette could increase the potential false positives (via spontaneous suppressor mutations) that would confound our analysis [30].
Thus, we used a plasmid shuffle strategy to determine whether cydC-D86G might represent a loss-of-function mutation. We generated strains that harbor the cydCD operon on a plasmid in a wild-type background, and separately, in a background with a precise deletion of the cydC locus (“Methods”). We then screened for spontaneous plasmid loss using the plasmid-borne counterselection gene sacB [31] (Fig. 2b). Wild-type cells lost a plasmid-borne copy of cydC at a rate five orders of magnitude higher than the ∆cydC strains, indicating that strains without functional cydC are not favored (Fig. 2b). As CydC is functional only when in a heterodimeric complex with its partner protein, CydD [26], our result also suggests that previous studies examining ∆cydC or ∆cydD mutants need to be interpreted with caution, and that mutant alleles thought to be total loss-of-function alleles may be partially active [23, 32]. We conclude from this result that cydC-D86G could not be encoding a nonfunctional protein in the strain isolated in our study, and that cydC-D86G necessarily retains the activity required for cell viability equivalent to the wild-type gene. Thus, cydC-D86G must be a gain-of-function mutant. This conclusion is also consistent with our previous result, which indicated that overexpression of cydC-D86G conferred tolerance to [EMIM]OAc, but overexpression of the wild-type cydC alone did not confer tolerance to the IL in this background (refer to Fig. 2a).
CydC does not improve IL tolerance via the cytochrome bd-I respiratory complex
Next, we sought to understand the mechanistic basis of IL tolerance in the E. coli cydC-D86G strain. The lack of a crystal structure for cydC makes it challenging to assess the role of a point mutation, especially in a transmembrane domain. One possible mechanism by which cydC could confer tolerance to [EMIM]OAc as well as improve terpene production is through the activation of the cytochrome AB complex, as some studies have implicated the cydCD gene product in assembling functional cytochrome bd-I (CydAB) complexes [22]. Cytochrome bd-I is also known to be involved in limiting oxidative damage such as that caused by the detoxification of free oxygen radicals [33]. Consistent with this notion, studies in budding yeast and filamentous fungi have implicated [EMIM]OAc in respiratory impairment [34,35,36]. If cytochrome CD complexes play a role in activating cytochrome bd-I, this could result in an indirect mechanism for [EMIM]OAc tolerance. Logically, the hypothesis based on these assumptions is that increased cytochrome bd-I activity would have the same behavior as the cydC-D86G mutant (Fig. 3a).

CydC Mediated [EMIM]OAc Tolerance Does Not Act Through cytochrome bd-I. a Schematic. Proposed relationship between CydC and cytochrome bd-I if [EMIM]OAc tolerance in CydC-D86G strains is due to directly activating cytochrome bd-I. b Conservation of cydC and cytochrome bd-I subunit cydB by strict (> 95% identity) homolog identification in bacteria. cydB is observed in a subset of species with detected cydC homologs. c Peroxide challenge assay. Wild-type DH1 cells or DH1 cydC-D86G cells grown in the presence of exogenous 30 mM hydrogen peroxide over a 20 h timeframe. No difference in hydrogen peroxide sensitivity was observed. Optical density (OD) is plotted on a log10 scale. d Tolerance to [EMIM]OAc in improved cytochrome bd-I cells. E. coli BW25113 ∆oppF [Ptrc-cydAB] or wild-type cells were grown in the presence of 50 mM [EMIM]OAc. Functional cytochrome bd-I activity was increased with 200 µM IPTG. Growth of cells was monitored over a 40 h timeframe. Optical density is plotted on a log10 scale
To evaluate this hypothesis, we used available genomic data and a phylogenetic approach to survey whether cydB and cydC always co-occur in bacterial genomes [37]. If CydC activity is strictly required to activate cytochrome bd-I (CydAB), it is likely that genomes with cydB would also encode cydC. This was not the case. Computational analysis identified highly conserved cydC homologs in 407 species, but only a subset of species (53 in total) contained a cydB homolog (Fig. 3b). Additionally, three isolates of E. coli encoded cydB, but did not contain a cydC homolog (Additional file 2: Table S1). These exceptions imply that cydC is not always required for cydB activity and furthermore, that cydC and cydB are not necessarily co-correlated in bacterial genomes.
The assumption that the function of cydC-D86G involves cytochrome bd-I activity also leads to two testable predictions. The first prediction is that E. coli cydC-D86G strains may have increased hydrogen peroxide resistance, the same phenotype as cells with increased cytochrome bd-I activity [38]. We subjected wild-type E. coli and E. coli cydC-D86G strains to increasing concentrations of exogenous hydrogen peroxide (“Methods”). We did not observe an increase in peroxide resistance in E. coli cydC-D86G cells, as both wild-type E. coli and E. coli cydC-D86G strains had the same sensitivity to exogenous hydrogen peroxide at concentrations up to 30 mM exogenous H2O2 (Fig. 3c). This observation is inconsistent with the hypothesis that cydC-D86G strains have more cytochrome bd-I complex activity. The second prediction is that strains with increased cytochrome bd-I activity may also have increased tolerance to the IL [EMIM]OAc. We tested this hypothesis using E. coli strains which we have previously shown to have increased cytochrome bd-I activity as measured by quantitatively improved hydrogen peroxide resistance [38]. This strain (E. coli ∆oppF PlacUV5-cydAB) was grown in the presence of [EMIM]OAc (Fig. 3d). We found that cydAB overexpression led to a longer doubling time and lower maximum growth (Fig. 3d). Induction of cydAB alone in the absence of the IL, or growth in the presence of the IL without induction of cydAB had no impact on growth (Additional file 1: Figure S3A, B). Additionally, we ruled out strain background effects by confirming that cydC and cydC-D86G confer IL tolerance in the related BW25113 strain background (Additional file 1: Figure S3C). The mutant protein CydC-D86G shows a function distinct from activating the cytochrome bd-I complex (Fig. 3a). Together these results indicate that strains with improved cytochrome bd-I activity are not tolerant to [EMIM]OAc, and do not recapitulate the IL tolerance that we observed in cydC-D86G strains.
Identification of a cydC-mediated response network independent of cytochrome bd-I activation
Since cydC-D86G is a gain-of-function mutant and acts independently from cytochrome bd-I activation, we speculated that this new activity was related to its role in maintaining cytoplasmic homeostasis and alleviating nitrosative stress [23]. With FT-IR and HPLC-LCMS analysis, we ruled out the possibility that cydC-D86G strains were consuming or modifying the [EMIM]+ ion (Additional file 1: Figure S4). To examine if the mutant cydC-D86G strain is more effective than the wild-type strain at upregulating global stress regulators in response to [EMIM]OAc, we took a systems-level approach to ascertain the affected downstream proteins in cydC-D86G cells.
Using shotgun proteomics, we compared samples with or without the cydC-D86G variant under typical production conditions (“Methods”) and measured the relative protein abundance levels for nearly 500 E. coli proteins. Samples from an empty vector control and the vector-borne cydC-D86G were harvested before treatment with [EMIM]OAc, 24 h after addition of the IL, and 48 h after addition of the IL. We identified proteins whose levels were increased or decreased at least twofold in the presence of [EMIM]OAc in cydC-D86G strains normalized to the protein levels before addition of [EMIM]OAc. The corresponding genes were binned into functional categories (COGs) and plotted against the overall gene abundances in the E. coli genome (Fig. 4a). Similar categorization was applied for all 486 proteins identified at high confidence to determine the selection bias from the analysis method compared to overall gene categories. We observed an enrichment of detected proteins related to “translation, ribosomal structure and biogenesis” (COG: J) when comparing total identified proteins to the E. coli genomic distribution. Due to their enrichment from the method alone, we considered these proteins to be false positives and did not pursue them further. However, we did detect enrichment of several gene categories that are specific to the increased IL tolerance in the cydC-D86G strains and which provide an improved understanding of IL tolerance in this mutant. Compared to wild-type cells and untreated cydC-D86G cells, genes in both “carbohydrate metabolism and transport (COG: G)” and “chaperone function, post translational modification, and protein turnover (COG: O)” categories were enriched when compared to the overall COG distribution relative to that in the wild type. The category “amino acid metabolism and transport (COG: E)” was also enriched in cydC-D86G strains treated with [EMIM]OAc, but in general the fold enrichment was higher in cydC-D86G strains then compared to the empty vector control. All identified genes and their enrichment values detected by this shotgun proteomics method are described in Additional file 3: Table S2.

Shotgun Proteomics Identifies Enriched Proteins in cydC-D86G Cells Upon [EMIM]OAc Treatment. a Strains harboring the limonene production plasmid (JBEI-6409) and a plasmid-borne copy of cydC-D86G [pTE42/Ptrc-cydD-cydC-D86G] were prepared for whole genome proteomic analysis (“Methods”). Samples were harvested immediately before induction with 1 µM IPTG and 48 h after induction. Proteins at least 2 × enriched were categorized into functional categories (COG, Cluster of Orthologous Groups) (“Methods”). White columns; distribution of COG terms from the E. coli genome [29]. Gray columns; subset of recovered proteins from shotgun proteomics binned into COGs and plotted as the percent of all proteins recovered. Blue striped bars; COG distribution from DH1 treated with 100 mM [EMIM]OAc plotted as the percent of enriched proteins. Black bars; COG distribution from DH1 cydC-D86G treated with 100 mM [EMIM]OAc plotted as the percent of enriched proteins. b Candidate effector proteins enriched at least 2× were validated with quantitative peptide-area proteomics. The same experimental regimen and cells used in a were used to confirm protein abundance. The fold enrichment (log2) of unique peptides are plotted against their normalized significance (P) values. COG descriptions. [C], Energy production and conversion. [E], Amino Acid metabolism and transport. [G], Carbohydrate metabolism and transport. [H], Coenzyme metabolism. [I], Lipid metabolism. [J], Translation. [K], Transcription. [M], Cell wall/membrane/envelope biogenesis. [O], Post-translational modification, protein turnover, chaperone function. [P]. Inorganic ion transport and metabolism. [S], Function Unknown. [T], Signal Transduction. [U], Intracellular Trafficking and Secretion
To validate these increased or decreased protein candidates, we developed an MRM based targeted proteomic method (“Methods”), and relatively quantified them among treatment conditions [39]. Statistical significance was confirmed across these four biological replicates for each treatment condition (Fig. 4b). The degree of peptide enrichment for both wild type and cydC-D86G strains was similar but proteins such as AdhP and DegP showed differential levels of enrichment (Fig. 4b). All proteins detected by the MRM method and their calculated p-values are described in Additional file 4: Table S3. Together, these results indicate that E. coli cells respond to [EMIM]OAc stress by upregulating amino acid metabolism related genes, but cydC-D86G strains specifically upregulate two additional gene classes, namely chaperones and carbohydrate metabolism genes.
To understand the role of these candidates as downstream effectors of the cydC-D86G phenotype, we reasoned that single gene deletion mutants might show differences in their ability to handle [EMIM]OAc stress. If a deletion mutant was sensitive to [EMIM]OAc, it would imply that the gene was required for the altered cydC-D86G stress response. Alternatively, deletion strains that were more resistant to the IL could indicate that cydC-D86G dampened the gene activity. However, a negative result, i.e. deletion mutants which had no change in phenotype, are also likely and are expected to arise from redundant or overlapping effector pathways.
We selected 20 deletion strains among protein candidates potentially involved in cydC-D86G mediated resistance to this IL. Many of the strains tested had subtle colony phenotypes compared to wild-type E. coli, ∆emrE, or ∆rcdA cells, two published deletion strains with sensitivity or resistance to [EMIM]OAc, respectively [16, 40]. Of the targets tested, E. coli ∆zapB cells were sensitive to this IL (Table 1). ∆adhP cells were more resistant to this IL, consistent with its statistically significant decrease in abundance in cydC-D86G strains (Table 1). ZapB is a non-essential protein which associates with FtsZ and is important for efficient chromosomal segregation [41]. The connection between increased ZapB activity and the role of cydCD complexes in this phenotype is unclear, but data for this candidate suggest that it may be a key response factor influenced by cydC activity in mediating tolerance to this IL. Essential genes such as plsB could not be examined. Overall, these results imply that while no single downstream gene is fully attributable for the cydC-D86G phenotype, specific categories of functions, chaperones, and carbohydrate metabolism can be implicated in the corresponding IL tolerance.
cydC-D86G strains produce high titers of limonene or isopentenol under [EMIM]OAc stress
As a cytochrome assembly factor or an amino acid transporter, cydC represents a different functional group from most tolerance mechanisms discovered to date. To test how well cydC-D86G strains produced limonene under [EMIM]OAc stress conditions, we cultivated cydC-D86G strains harboring the limonene production plasmid (JBEI-6409) [42] in the presence of 100 mM [EMIM]OAc. Strains harboring either wild-type cydC or cydC-D86G at the cydC genomic locus were examined for limonene production. Samples were collected 24 and 48 h after induction of the limonene production pathway with 15 µM IPTG. We observed that, unlike previously discovered IL tolerant strains, the cydC-D86G strain was an excellent limonene producer in the presence of 100 mM [EMIM]OAc. The cydC-D86G strain reached titers of above 200 mg/L limonene in 48 h, and a specific production of 80 mg/L/OD (Fig. 5a, Additional file 1: Figure S5), equivalent to the limonene production in the absence of IL in any of the strains tested. In contrast, the ∆rcdA strain only partially restored production under these cultivation parameters (Fig. 5a). Wild-type E. coli cells carrying only the limonene production plasmid, or an additional copy of wild-type cydC failed to grow in the presence of [EMIM]OAc and could not be tested for limonene production (Fig. 5a). We conclude from these results that the cydC-D86G strain has two advantages: it increases tolerance to exogenous [EMIM]OAc and restores high titers of limonene in the presence of this IL.

Production of the Biofuel Precursor Limonene and Isopentenol Under Exogenous [EMIM]OAc Growth Conditions. a E. coli DH1 cells with or without the cydC-D86G mutation at the genomic locus and a limonene production plasmid (JBEI-6409) were tested for limonene production in the presence or absence of 100 mM [EMIM]OAc (“Methods”). Time points were sampled 24 h or 48 h after pathway induction with 15 µM IPTG. Black bars; WT strains. Grey bars; cydC-D86G strains. Orange bars; ∆rcdA strains. b E. coli DH1 ∆pta∆poxB∆ackA (JBEI-3606) harboring [pTE50/PcydD-cydD-cydC] (white bars), [pTE88/PcydD-cydD-cydC-D86G] (red striped bars), or [pTE100 PcydD-cydD-cydC-D86V] (blue bars) along with an isopentenol production plasmid (JBEI-9321) were prepared for biofuel production in the presence of 75 mM [EMIM]OAc. Sampling was conducted at 24 h or 48 h after pathway induction with 500 µM IPTG. Production of isopentenol was compared to E. coli DH1 strains harboring an empty plasmid (PcydD only) and the isopentenol production plasmid (JBEI-9321) in the absence of [EMIM]OAc. Only trace isopentenol production (with high acetate formation) was observed in the presence of [EMIM]OAc using either E. coli DH1 or E. coli DH1 cydC-D86G strains (data not shown)
To examine if the improvement in production was specific to limonene as a final product, we tested the production of another biofuel candidate and platform chemical derived from the mevalonate pathway, 3-methyl-3-buten-1-ol, (hereafter referred to as isopentenol) [43, 44]. When we assayed the initial isopentenol production strain which carried both the isopentenol production pathway as well as cydC-D86G on a second plasmid, only trace amounts of isopentenol were detected by GC–FID when cultured in the presence of exogenous [EMIM]OAc. Instead, the majority of the product was acetate. To address a potential shift in metabolic flux towards acetate, we used acetate-route deficient E. coli cells, which lack the three major acetate pathway operons, ∆ackA ∆poxB ∆pta [45]. Exploiting our new plasmid-based cydC system, we examined E. coli ∆ackA ∆poxB ∆pta cells harboring both the isopentenol production pathway and plasmids with wild-type cydC, cydC-D86G or cydC-D86V. Similar to the results with limonene production in the presence of [EMIM]OAc, cells carrying wild-type cydC plasmid grew very poorly; we detected poor production of isopentenol in those cultures. In contrast, cells carrying the cydC-D86G plasmid produced robust titers of isopentenol in the presence of [EMIM]OAc (Fig. 5b), similar to published isopentenol production titers using this pathway in the absence of ILs [44]. Production titers were even higher in strains carrying a cydC-D86V plasmid, emphasizing the portability of this mutation to different strain backgrounds (Fig. 5b). From these results we conclude that cydC-D86G and cydC-D86V confer the robust capacity to generate two final products (limonene and isopentenol) under [EMIM]OAc stress, which to our knowledge is a first demonstration in the field.
Discussion
ILs are a double-edged sword as a biomass pretreatment reagent: they are appealing because they can help efficiently extract sugar from recalcitrant lignocellulosic plant biomass, but often pose a detrimental effect on the downstream enzymes and microbial hosts for bioproduction of a target compound. In earlier reports, the improvement in IL tolerance has been shown to enhance product titers relative to strain productivity before engineering, but not at the levels possible under optimal growth conditions [14, 16, 46, 47]. This inability to fully recover production levels after engineering strains for IL tolerance indicates that the IL impacts cell growth and biosynthetic pathways separately; the biosynthetic pathway was still affected, even though overall growth under these conditions was restored. In combination, even minor reductions in strain productivity would limit the development of one-pot processes or even the use of unwashed pretreated biomass. The E. coli cydC-D86G chassis is an important step forward in the field, as it is both IL tolerant and retains the optimal efficiency of producing advanced biofuels. Future processes could utilize this mutation in cydC to develop one-pot methods with [EMIM]OAc pretreated lignocellulosic biomass, without compromising biofuel production.
We could not find the exact mechanism for the gain of function activity of the mutant cydC-D86G because cydC encodes an essential gene, a gene category that is challenging to utilize in metabolic engineering studies. Conditionally-essential gene collections are not yet widespread in bacteria, as has been completed for budding yeast [48]. The development of inducible CRISPRi libraries [49] will facilitate utilization of this gene reservoir and elucidate their cellular functions [50]. Spontaneous mutagenesis screens, such as via laboratory evolution, remain the standard for identifying advantageous gain-of-function mutations.
As evidenced by the breadth of functional categories implicated by our proteomics data, ionic liquids have a pleiotropic impact on bacterial systems. Similar results have been reported in other model organisms: in the yeast S. cerevisiae, [EMIM]OAc has been reported to cause a growth impact that exceeds that of [EMIM]Cl or NaOAc [51], and is thought to deform cell wall structure and limit oxygen transfer from the culture media into cells [35]. While we did not detect a decrease of the expression of genes related to fatty acid synthesis from this analysis, we did detect a five-fold decrease in the abundance of an essential gene, plsB (see Additional file 3: Table S2). PlsB is a critical for the downstream selection of fatty acids incorporated into membrane phospholipids, which could impact cell membrane structure or integrity [52]. However, the relationship between membrane lipid and protein composition and cydC-D86G remains to be unraveled.
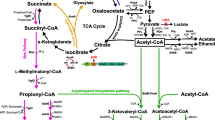
Previous studies have primarily resulted in the identification of native and non-native efflux pumps whose expression confers tolerance to [EMIM]OAc [14, 16, 53, 54]. In this study, we identify a new mechanism for bacterial resistance to [EMIM]OAc that addresses both the IL toxicity impact on cell growth and also protects the production pathway from inhibitory impact. It is unclear why mutations in candidates from two different protein families, specifically, efflux pumps and CydCD complex both conferred tolerance towards IL. However, both categories are fairly ubiquitous across bacteria and have broad substrate specificity [55]. We have demonstrated that our mutant, cydC-D86G, is a gain-of-function mutant and the over-expression of wild-type cydC alone does not confer tolerance to this IL in E. coli. While the complete mechanism for this IL tolerance remains to be understood, we speculate that this point mutation renders CydCD-complexes more promiscuous or active, but retains the minimum activities required for cellular viability. The mutant allele of cydC could result in CydCD-complexes with additional capacity to enhance multi-genic functions in carbohydrate transport and protein turnover/post translational modifications, without necessarily exporting a toxic metabolite from the cytoplasm (see Fig. 6). These gross changes could result in a distinct cytosolic response, blocking both the downstream impacts [EMIM]OAc on cellular processes as well as protecting the cell from the burden imposed by the mevalonate pathway for limonene or isopentenol production.

CydC modulates many cellular activities. Proposed role of cydC and ionic liquid tolerance. Proteomic analysis and experimental validation in E. coli deletion strains implicate a suite of pathways involved in the cydC response to [EMIM]OAc. In the top panel, cells express a heterologous gene pathway to produce a desired product, turning sugar (blue hexagon) into the target compound (green hexagon). The addition of the IL [EMIM]OAc in the middle panel has a broad impact on cellular physiology, impacting respiration, inorganic ion response factors, transporters, which ultimately inhibit cell growth and desired production. However, the cydC-D86G mutant in the bottom panel has a distinct proteomic response, and instead upregulates unique gene function categories, such as [U] intracellular trafficking and secretion and [G] carbohydrate metabolism and transport (see Fig. 4 for complete legend), ultimately restoring both cell growth and desired biofuel production
Our strategy to develop a microbial host chassis for the production of limonene in the presence of the IL, [EMIM]OAc, provides a template to obtain gain-of-function mutants applicable for cultivation under industrially-relevant conditions. By closely mimicking the conditions under which the target compound is produced, we obtained a single gain-of-function mutant in five independent trials in less than 40 h of incubation. In contrast to other laboratory evolution studies [15, 56, 57], we used an inhibitory concentration of the IL and included the potential burden of the limonene production plasmid in the screen. Future studies should consider the burden of a heterologous gene pathway as a fitness disadvantage when undertaking evolution studies.
This study exemplifies the use of interdisciplinary approaches to classical genetics and microbiology in conjunction with rational strain engineering to provide an elegant blueprint for optimizing microbial biofuel production under otherwise unfavorable culturing conditions. Heterologous gene pathways are a metabolic burden and have idiosyncratic impacts on native microbial metabolism. While cydC-D86G strains were already competent to produce d-limonene, the production of isopentenol in the presence of exogenous [EMIM]OAc required the use of acetate deficient strains to achieve the highest titer of isopentenol under these stress conditions. In retrospect, it is clear that a heterologous gene pathway could impact the native host metabolism, as we observed when we initially sought to produce isopentenol under IL stress conditions. A unified strategy for engineering microbial hosts will be critical in the development of promising non-model microorganisms in one-pot processes when final product toxicity is inhibitory [58, 59]. Metabolic flux models which incorporate data from all of these relevant impediments on central metabolism will advance our efforts to build robust cell factories.
Methods
Reagents, bacterial strains and plasmids
All chemicals used were reagent grade and purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise noted. [EMIM]OAc, also known as [C2C1im]OAc, was purchased from Sigma-Aldrich and used as received. All strains and plasmids used in this work are listed at http://public-registry.jbei.org and are summarized in Additional file 5: Table S4. E. coli DH10B (Invitrogen, Carlsbad, CA) was used for cloning and manipulation of plasmids. Except for the experiments described in Table 1 from the Keio Collection, all IL tolerance assays were conducted with E. coli DH1. Mutants from the Keio collection were tested against the BW25113 wild-type strain. Plasmids were constructed using isothermal assembly with 40 bp overlapping homologous sequences and plasmid sequences were verified with restriction endonuclease digestion and Sanger Sequencing (Quintara Biosciences, Albany, CA). We defined the promoter sequence of the cydDC operon as the immediate upstream ~ 500 bp of cydC. Plasmid transformation was conducted as previously described [60] or else as expressly indicated.
Generation of spontaneous mutants tolerant to [EMIM]OAc
Escherichia coli DH1 strains with or without specific genomic mutations and a limonene production plasmid were struck out from glycerol stocks and single colonies were used to inoculate 5 mL LB precultures. These individual precultures were then back-diluted into synthetic culture media with a 20% dodecane (Sigma Aldrich, St. Louis, MO) overlay and incubated at 30 °C until the cultures showed evidence of growth.
[EMIM]OAc tolerant E. coli strains were examined to determine if they contained heritable mutations as follows. Recovered mutants were washed three times in sterile deionized water and resuspended in fresh EZ Rich 1% d-glucose media. Approximately 100 µL of this resuspension was used to reinnoculate a fresh 25 mL culture lacking any ionic liquid. The optical density was measured over a 16-h period to ensure that at least eight doublings had occurred. Cells were then washed with sterile water as before and a similar back dilution into EZ Rich 1% d-glucose containing 100 mM [EMIM]OAc. The optical density was measured as before.
Identification of mutant alleles by next generation illumina sequencing
Genomic DNA from candidate mutant strains was isolated using standard methods, and libraries were prepared for Illumina sequencing at the Joint Genome Institute (Walnut creek, CA, USA) as described by the manufacturer. In brief, DNA was randomly sheared into ~ 500 bp fragments to create an Illumina library. This library was sequenced on Illumina Miseq generating 150 bp paired end reads. Reads were aligned to the reference genome using BWA and putative SNPs and small indels were called using samtools and mpileup [61, 62]. Putative structural variants were called using a combination of BreakDancer (filtered to quality 90+), and Pindel [63, 64]. Basecall variants were called with CONV essentially as described by [65]. Candidate mutations were verified as absent in the parental E. coli genomic strain by Sanger Sequencing (Quintara Biosciences, South San Francisco CA).
Strain culturing conditions
Limonene and isopentenol production
All production assays were conducted in the E. coli DH1 strain background. Strains containing the limonene production plasmid (JBEI-6409) were grown in EZ-Rich medium with the appropriate antibiotics to maintain plasmid selection, with glucose as the carbon source at a final concentration of 1% (10 g/L) (Teknova, Hollister, CA). 5 mL precultures were grown shaking (200 rpm) overnight at 30 °C and were used to inoculate starting 25 mL production cultures at an optical density of ~ 0.1 OD600. Cultures were grown for two doublings, to an OD600 of 0.4–0.6, at which expression of limonene production genes was induced with 15 μM β-d-1-thiogalactopyranoside (IPTG, Sigma Aldrich, St. Louis MO). A dodecane (Sigma Aldrich, St. Louis, MO) overlay (5 mL for 25 mL cultures) was applied at the time of IPTG induction, and was used to extract biosynthetically produced d-limonene. Limonene production was quantified 24 h or 48 h after IPTG induction using GC–MS as previously described [42].
The production of isopentenol was carried out essentially as described in Kang et al. [44] using the isopentenyl diphosphate bypass pathway to generate isopentenol. Briefly, cells of the indicated genotype were grown in EZ-Rich medium with the appropriate antibiotics to maintain plasmid selection, with glucose at the final concentration of 1% (10 g/L). Isopentenol production genes were induced with 500 μM IPTG. No dodecane overlay was applied. Isopentenol production was quantified 24 h or 48 h after IPTG induction using either GC–MS or GC–FID (Flame Ionization Detector) as previously described [43, 44].
Preparation of cells for proteomic analysis
To measure global protein abundance changes under IL stress conditions, the same strains used for limonene production in Fig. 4a were prepared exactly as described in 1% glucose EZ-Rich media, induced with 15 μM IPTG and a 20% dodecane overlay, in a 25 mL culture volume. Approximately 3 × 109 cells were harvested by centrifugation at 14,000g for 3 min and stored at − 80 °C until analysis. Four biological replicates across two trials were used for the shotgun proteomics analysis. Another 3 × 109 cells were harvested by centrifugation and prepared for peptide analysis. Samples for peptide analysis were also prepared in biological quadruplicate.
Assessment of gene essentiality by plasmid shuffle
Wild-type E. coli DH1 strains were sequentially transformed with pKD46 [Pbad-gam-beta-exo AmpR] followed by pTE50 [sacB PCYDD-cydDC KanR] using electroporation (Protocol No. 26, page 1.119) [29, 66]. Briefly, a fresh colony from a glycerol stock was inoculated into a 50 mL LB 1% arabinose (Sigma-Aldrich, St. Louis MO) preculture, and grown to an optical density of ~ 0.4 at 600 nm. Cells were harvested by centrifugation at 4000g for 10 min, washed three times with 50 mL cold dH2O, and prepared for transformation via electroporation by resuspending the washed cell pellet in 1 mL of 10% glycerol. Cells were transformed with approximately 100 ng of a PCR fragment containing the Hygromycin B drug cassette [67] flanked on either side with 3 kb of homology to the upstream or downstream regions immediately adjacent to cydC. After DNA electroporation, cells were outgrown for 3 h in SOC media (New England Biolabs, Ipswich, MA) supplemented with 0.5% arabinose and plated onto LB plates containing 70 µg/mL Hygromycin B (Roche Life Sciences, Indianapolis IN). Hygromycin resistant clones were observed after 1 day post plating after incubation at 30 °C. Genomic deletions were verified by colony PCR to ensure replacement of the cydC locus with the hygromycin cassette using primers flanking the deletion cassette that would not amplify the plasmid-borne cydC. Spontaneous loss of pKD46 was confirmed after incubating validated clones at 42 °C for 1 day on LB plates and testing single colonies for the ability to grow on LB ampicillin plates. Due to the extended outgrowth phase used in this protocol, three independent electroporation reactions were conducted in parallel, and multiple validated colonies from different transformations were used for subsequent experiments.
Following strain generation, strains harboring either the wild-type cydC locus or the genomic deletion as well as pTE50 [sacB PCYDD-cydDC KanR] were struck to single colonies from glycerol stocks. Single colonies were inoculated into LB precultures for 24 h to allow for spontaneous plasmid loss in the population. After outgrowth, cells were serially diluted in a 96 well microtiter plate and 3 μL of each tenfold dilution was plated on solid agar media containing LB supplemented with or without 10% sucrose. Photomicrographs of each plate were taken approximately 1 day post plating.
Peroxide sensitivity assay
To determine the correlation between [EMIM]OAc resistance and hydrogen peroxide resistance, candidate deletion strains from the E. coli BW25113 background isolated from the Keio collection were selected for analysis [29]. Genomic deletions were confirmed by colony PCR using primers flanking the kanamycin drug cassette and the hydrogen peroxide assay was conducted as previously described [68]. In brief, fresh overnight cultures of the appropriate genotype were back diluted into 96 well microtiter dishes containing LB media supplemented with or without 30 mM hydrogen peroxide (Sigma Aldrich, St. Louis, MO). Growth was monitored in a BioTek (Winooski, VT) Synergy plate reader by measuring optical density at OD600 at 15-min intervals. All experiments were conducted in triplicate and repeated at least twice over the course of several weeks.
Proteomics
Samples prepared for shotgun proteomic experiments were analyzed by using an Agilent 6550 iFunnel Q-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA) coupled to an Agilent 1290 UHPLC system as described previously [69]. Twenty (20) μg of peptides were separated on a Sigma–Aldrich Ascentis Peptides ES-C18 column (2.1 mm × 100 mm, 2.7 μm particle size, operated at 60 °C) at a 0.400 mL/min flow rate and eluted with the following gradient described previously [69]. The acquired data was exported as MGF files and searched against the latest E. coli (strain K12) protein database with Mascot search engine version 2.3.02 (Matrix Science). The resulting search results were filtered and analyzed by Scaffold v 4.3.0 (Proteome Software Inc.). Normalized spectra counts of identified proteins were exported for quantitative analysis and selection of target proteins, which were significantly altered between experimental groups. These significantly altered proteins were classified into COG terms using eggnog 4.5 and are accessible at https://goo.gl/GgQGns.
A targeted SRM method was developed to target peptides of these proteins by using an in-house build E. coli spectral library. The SRM targeted proteomic assays were performed, as described previously [39], on an Agilent 6460 QQQ mass spectrometer system coupled with an Agilent 1290 UHPLC system (Agilent Technologies, Santa Clara, CA). Peptides were separated on an Ascentis Express Peptide C18 column [2.7-mm particle size, 160-Å pore size, 5-cm length × 2.1-mm inside diameter (ID), coupled to a 5-mm × 2.1-mm ID guard column with the same particle and pore size, operating at 60 °C; Sigma-Aldrich] operating at a flow rate of 0.4 mL/min via the following gradient: initial conditions were 98% solvent A (0.1% formic acid), 2% solvent B (99.9% acetonitrile, 0.1% formic acid). Solvent B was increased to 35% over 6.5 min, and was then increased to 80% over 1.5 min, and held for 1.5 min at a flow rate of 0.6 mL/min, followed by a ramp back down to 2% B over 0.5 min where it was held for 1 min to re-equilibrate the column to original conditions. The data were acquired using Agilent MassHunter version B.08.02. Acquired SRM data were analyzed by Skyline software version 3.70 (MacCoss Lab Software). The SRM methods and data are available at Panoramaweb [70] https://goo.gl/GgQGns.
Quantification of [EMIM]OAc in growth media by fourier-transform infrared spectroscopy (FT-IR) and high performance liquid chromatography–liquid chromatography mass spectrometry (HPLC–LCMS)
Quantification of [EMIM]OAc by FTIR was done essentially as described in [71] using cells grown in LB media. Briefly, quantitative measurement of the imidazolium-based ionic liquid was interrogated using the unique peak height at 1170.59 cm−1 corresponding to the presence of [EMIM]OAc. A standard curve was generated using commercially synthesized [EMIM]OAc. LC–MS analysis of [EMIM]+ was carried out with a method similar to the one described by Bokinsky et al. that was used to measure amino acids [72]. The only differences were the column compartment temperature, which was set to 20 °C, and the LC gradient elution method, described as follows: linearly increased from 90% B to 70% B in 4 min, held at 70% B for 1.5 min, decreased from 70% B to 40% B in 0.5 min, held at 40% B for 2.5 min, increased from 40% B to 90% B in 0.5 min, and held at 90% B for 2 min. The flow rate was held at 0.6 mL/min for 6.5 min, increased from 0.6 mL/min to 1 mL/min in 0.5 min, and held at 1 mL/min for 4 min. The total LC run time was 11 min.
Abbreviations
- IL:
-
ionic liquid
- [EMIM]OAc and [C2C1im]OAc:
-
1-ethyl-3-methyl-imidizolium acetate
- IPTG:
-
isopropyl β-d-1-thiogalactopyranoside
- COG:
-
clusters of orthologous groups
- FID:
-
flame ionization detector
References
Humbird D, Davis R, Tao L, Kinchin C, Hsu D, Aden A, Schoen P, Lukas J, Olthof B, Worley M. Process design and economics for biochemical conversion of lignocellulosic biomass to ethanol: dilute-acid pretreatment and enzymatic hydrolysis of corn stover. Golden: National Renewable Energy Lab (NREL); 2011.
Mosier N, Wyman C, Dale B, Elander R, Lee Y, Holtzapple M, Ladisch M. Features of promising technologies for pretreatment of lignocellulosic biomass. Biores Technol. 2005;96:673–86.
Alvira P, Tomás-Pejó E, Ballesteros M, Negro MJ. Pretreatment technologies for an efficient bioethanol production process based on enzymatic hydrolysis: a review. Biores Technol. 2010;101:4851–61.
Cornmell RJ, Winder CL, Tiddy GJT, Goodacre R, Stephens G. Accumulation of ionic liquids in Escherichia coli cells. Green Chem. 2008;10:836–41.
Pernak J, Sobaszkiewicz K, Mirska I. Anti-microbial activities of ionic liquids. Green Chem. 2003;5:52–6.
Ventura SP, de Barros RL, Sintra T, Soares CM, Lima AS, Coutinho JA. Simple screening method to identify toxic/non-toxic ionic liquids: agar diffusion test adaptation. Ecotoxicol Environ Saf. 2012;83:55–62.
Konda NM, Shi J, Singh S, Blanch HW, Simmons BA, Klein-Marcuschamer D. Understanding cost drivers and economic potential of two variants of ionic liquid pretreatment for cellulosic biofuel production. Biotechnol Biofuels. 2014;7:86.
Mukhopadhyay A. Tolerance engineering in bacteria for the production of advanced biofuels and chemicals. Trends Microbiol. 2015;23:498–508.
Ranke J, Muller A, Bottin-Weber U, Stock F, Stolte S, Arning J, Stormann R, Jastorff B. Lipophilicity parameters for ionic liquid cations and their correlation to in vitro cytotoxicity. Ecotoxicol Environ Saf. 2007;67:430–8.
Ventura SP, Goncalves AM, Sintra T, Pereira JL, Goncalves F, Coutinho JA. Designing ionic liquids: the chemical structure role in the toxicity. Ecotoxicology. 2013;22:1–12.
Wang S, Zhao W, Lee TS, Singer SW, Simmons BA, Singh S, Yuan Q, Cheng G. Dimethyl sulfoxide assisted ionic liquid pretreatment of switchgrass for isoprenol production. ACS Sustain Chem Eng. 2018;6:4354–61.
Yamada R, Nakashima K, Asai-Nakashima N, Tokuhara W, Ishida N, Katahira S, Kamiya N, Ogino C, Kondo A. Direct ethanol production from ionic liquid-pretreated lignocellulosic biomass by cellulase-displaying yeasts. Appl Biochem Biotechnol. 2017;182:229–37.
Peralta-Yahya PP, Ouellet M, Chan R, Mukhopadhyay A, Keasling JD, Lee TS. Identification and microbial production of a terpene-based advanced biofuel. Nat Commun. 2011;2:483.
Ruegg TL, Kim EM, Simmons BA, Keasling JD, Singer SW, Lee TS, Thelen MP. An auto-inducible mechanism for ionic liquid resistance in microbial biofuel production. Nat Commun. 2014;5:3490.
Mohamed ET, Wang S, Lennen RM, Herrgard MJ, Simmons BA, Singer SW, Feist AM. Generation of a platform strain for ionic liquid tolerance using adaptive laboratory evolution. Microb Cell Fact. 2017;16:204.
Frederix M, Mingardon F, Hu M, Sun N, Pray T, Singh S, Simmons BA, Keasling JD, Mukhopadhyay A. Development of an E. coli strain for one-pot biofuel production from ionic liquid pretreated cellulose and switchgrass. Green Chem. 2016;18:4189–97.
Mora-Pale M, Meli L, Doherty TV, Linhardt RJ, Dordick JS. Room temperature ionic liquids as emerging solvents for the pretreatment of lignocellulosic biomass. Biotechnol Bioeng. 2011;108:1229–45.
Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. The cytochrome bd respiratory oxygen reductases. Biochim Biophys Acta. 2011;1807:1398–413.
Li C, Tanjore D, He W, Wong J, Gardner JL, Sale KL, Simmons BA, Singh S. Scale-up and evaluation of high solid ionic liquid pretreatment and enzymatic hydrolysis of switchgrass. Biotechnol Biofuels. 2013;6:154.
Mans R, Daran JG, Pronk JT. Under pressure: evolutionary engineering of yeast strains for improved performance in fuels and chemicals production. Curr Opin Biotechnol. 2018;50:47–56.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–84.
Georgiou CD, Fang H, Gennis RB. Identification of the cydC locus required for expression of the functional form of the cytochrome d terminal oxidase complex in Escherichia coli. J Bacteriol. 1987;169:2107–12.
Holyoake LV, Hunt S, Sanguinetti G, Cook GM, Howard MJ, Rowe ML, Poole RK, Shepherd M. CydDC-mediated reductant export in Escherichia coli controls the transcriptional wiring of energy metabolism and combats nitrosative stress. Biochem J. 2016;473:693–701.
Pittman MS, Robinson HC, Poole RK. A bacterial glutathione transporter (Escherichia coli CydDC) exports reductant to the periplasm. J Biol Chem. 2005;280:32254–61.
Shepherd M. The CydDC ABC transporter of Escherichia coli: new roles for a reductant efflux pump. Biochem Soc Trans. 2015;43:908–12.
Yamashita M, Shepherd M, Booth WI, Xie H, Postis V, Nyathi Y, Tzokov SB, Poole RK, Baldwin SA, Bullough PA. Structure and function of the bacterial heterodimeric ABC transporter CydDC: stimulation of ATPase activity by thiol and heme compounds. J Biol Chem. 2014;289:23177–88.
Goldman BS, Gabbert KK, Kranz RG. The temperature-sensitive growth and survival phenotypes of Escherichia coli cydDC and cydAB strains are due to deficiencies in cytochrome bd and are corrected by exogenous catalase and reducing agents. J Bacteriol. 1996;178:6348–51.
Poole RK, Gibson F, Wu G. The cydD gene product, component of a heterodimeric ABC transporter, is required for assembly of periplasmic cytochrome c and of cytochrome bd in Escherichia coli. FEMS Microbiol Lett. 1994;117:217–23.
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2006(2):0008.
Sikorski RS, Boeke JD. In vitro mutagenesis and plasmid shuffling: from cloned gene to mutant yeast. Methods Enzymol. 1991;194:302–18.
Pelicic V, Reyrat JM, Gicquel B. Expression of the Bacillus subtilis sacB gene confers sucrose sensitivity on mycobacteria. J Bacteriol. 1996;178:1197–9.
Cruz-Ramos H, Cook GM, Wu G, Cleeter MW, Poole RK. Membrane topology and mutational analysis of Escherichia coli CydDC, an ABC-type cysteine exporter required for cytochrome assembly. Microbiology. 2004;150:3415–27.
Lindqvist A, Membrillo-Hernandez J, Poole RK, Cook GM. Roles of respiratory oxidases in protecting Escherichia coli K12 from oxidative stress. Antonie Van Leeuwenhoek. 2000;78:23–31.
Dickinson Q, Bottoms S, Hinchman L, McIlwain S, Li S, Myers CL, Boone C, Coon JJ, Hebert A, Sato TK, et al. Mechanism of imidazolium ionic liquids toxicity in Saccharomyces cerevisiae and rational engineering of a tolerant, xylose-fermenting strain. Microb Cell Fact. 2016;15:17.
Mehmood N, Husson E, Jacquard C, Wewetzer S, Buchs J, Sarazin C, Gosselin I. Impact of two ionic liquids, 1-ethyl-3-methylimidazolium acetate and 1-ethyl-3-methylimidazolium methylphosphonate, on Saccharomyces cerevisiae: metabolic, physiologic, and morphological investigations. Biotechnol Biofuels. 2015;8:17.
Singer SW, Reddy AP, Gladden JM, Guo H, Hazen TC, Simmons BA, VanderGheynst JS. Enrichment, isolation and characterization of fungi tolerant to 1-ethyl-3-methylimidazolium acetate. J Appl Microbiol. 2011;110:1023–31.
Dehal PS, Joachimiak MP, Price MN, Bates JT, Baumohl JK, Chivian D, Friedland GD, Huang KH, Keller K, Novichkov PS, et al. MicrobesOnline: an integrated portal for comparative and functional genomics. Nucleic Acids Res. 2010;38:D396–400.
Jensen HM, Eng T, Chubukov V, Herbert RA, Mukhopadhyay A. Improving membrane protein expression and function using genomic edits. Sci Rep. 2017;7:13030.
Batth TS, Singh P, Ramakrishnan VR, Sousa MML, Chan LJG, Tran HM, Luning EG, Pan EHY, Vuu KM, Keasling JD, et al. A targeted proteomics toolkit for high-throughput absolute quantification of Escherichia coli proteins. Metab Eng. 2014;26:48–56.
Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, Blow MJ, Bristow J, Butland G, Arkin AP, Deutschbauer A. Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. MBio. 2015;6:e00306–15.
Ebersbach G, Galli E, Moller-Jensen J, Lowe J, Gerdes K. Novel coiled-coil cell division factor ZapB stimulates Z ring assembly and cell division. Mol Microbiol. 2008;68:720–35.
Alonso-Gutierrez J, Chan R, Batth TS, Adams PD, Keasling JD, Petzold CJ, Lee TS. Metabolic engineering of Escherichia coli for limonene and perillyl alcohol production. Metab Eng. 2013;19:33–41.
George KW, Thompson MG, Kang A, Baidoo E, Wang G, Chan LJ, Adams PD, Petzold CJ, Keasling JD, Lee TS. Metabolic engineering for the high-yield production of isoprenoid-based C(5) alcohols in E. coli. Sci Rep. 2015;5:11128.
Kang A, George KW, Wang G, Baidoo E, Keasling JD, Lee TS. Isopentenyl diphosphate (IPP)-bypass mevalonate pathways for isopentenol production. Metab Eng. 2016;34:25–35.
Dittrich CR, Bennett GN, San KY. Characterization of the acetate-producing pathways in Escherichia coli. Biotechnol Prog. 2005;21:1062–7.
Bokinsky G, Peralta-Yahya PP, George A, Holmes BM, Steen EJ, Dietrich J, Lee TS, Tullman-Ercek D, Voigt CA, Simmons BA, Keasling JD. Synthesis of three advanced biofuels from ionic liquid-pretreated switchgrass using engineered Escherichia coli. Proc Natl Acad Sci USA. 2011;108:19949–54.
Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJ, Hanai T, Liao JC. Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng. 2008;10:305–11.
Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart-Ornstein J, Newman HW, Braun S, Madhani HD, Krogan NJ, Weissman JS. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 2008;5:711–8.
Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo BM, Marta E, et al. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell. 2016;165:1493–506.
Juhas M, Eberl L, Church GM. Essential genes as antimicrobial targets and cornerstones of synthetic biology. Trends Biotechnol. 2012;30:601–7.
Ouellet M, Datta S, Dibble DC, Tamrakar PR, Benke PI, Li C, Singh S, Sale KL, Adams PD, Keasling JD, et al. Impact of ionic liquid pretreated plant biomass on Saccharomyces cerevisiae growth and biofuel production. Green Chem. 2011;13:2743–9.
Heath RJ, Jackowski S, Rock CO. Guanosine tetraphosphate inhibition of fatty acid and phospholipid synthesis in Escherichia coli is relieved by overexpression of glycerol-3-phosphate acyltransferase (plsB). J Biol Chem. 1994;269:26584–90.
Frederix M, Hutter K, Leu J, Batth TS, Turner WJ, Ruegg TL, Blanch HW, Simmons BA, Adams PD, Keasling JD, et al. Development of a native Escherichia coli induction system for ionic liquid tolerance. PLoS ONE. 2014;9:e101115.
Higgins DA, Young MK, Tremaine M, Sardi M, Fletcher JM, Agnew M, Liu L, Dickinson Q, Peris D, Wrobel RL, et al. Natural variation in the multidrug efflux pump SGE1 underlies ionic liquid tolerance in yeast. Genetics. 2018. https://doi.org/10.1534/genetics.118.301161.
Puri N, Prakash O, Manoharlal R, Sharma M, Ghosh I, Prasad R. Analysis of physico-chemical properties of substrates of ABC and MFS multidrug transporters of pathogenic Candida albicans. Eur J Med Chem. 2010;45:4813–26.
Reider Apel A, Ouellet M, Szmidt-Middleton H, Keasling JD, Mukhopadhyay A. Evolved hexose transporter enhances xylose uptake and glucose/xylose co-utilization in Saccharomyces cerevisiae. Sci Rep. 2016;6:19512.
Van Hofwegen DJ, Hovde CJ, Minnich SA. Rapid evolution of citrate utilization by Escherichia coli by direct selection requires citT and dctA. J Bacteriol. 2016;198:1022–34.
Sundstrom E, Yaegashi J, Yan J, Masson F, Papa G, Rodriguez A, Mirsiaghi M, Liang L, He Q, Tanjore D. Demonstrating a separation-free process coupling ionic liquid pretreatment, saccharification, and fermentation with Rhodosporidium toruloides to produce advanced biofuels. Green Chem. 2018;20:2870–9.
Rodriguez A, Salvachúa D, Katahira R, Black BA, Cleveland NS, Reed M, Smith H, Baidoo EEK, Keasling JD, Simmons BA, et al. Base-catalyzed depolymerization of solid lignin-rich streams enables microbial conversion. ACS Sustain Chem Eng. 2017;5:8171–80.
Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci USA. 1989;86:2172–5.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome project data processing S: the sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–71.
Le Crom S, Schackwitz W, Pennacchio L, Magnuson JK, Culley DE, Collett JR, Martin J, Druzhinina IS, Mathis H, Monot F, et al. Tracking the roots of cellulase hyperproduction by the fungus Trichoderma reesei using massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:16151–6.
Sambrook J, Russell DW. Transformation of E coli by electroporation. Cold Spring Harb Protoc. 2006. https://doi.org/10.1101/pdb.prot3933.
Kaster KR, Burgett SG, Rao RN, Ingolia TD. Analysis of a bacterial hygromycin B resistance gene by transcriptional and translational fusions and by DNA sequencing. Nucleic Acids Res. 1983;11:6895–911.
Jensen ED, Ferreira R, Jakociunas T, Arsovska D, Zhang J, Ding L, Smith JD, David F, Nielsen J, Jensen MK, Keasling JD. Transcriptional reprogramming in yeast using dCas9 and combinatorial gRNA strategies. Microb Cell Fact. 2017;16:46.
Gonzalez Fernandez-Nino SM, Smith-Moritz AM, Chan LJ, Adams PD, Heazlewood JL, Petzold CJ. Standard flow liquid chromatography for shotgun proteomics in bioenergy research. Front Bioeng Biotechnol. 2015;3:44.
Sharma V, Eckels J, Taylor GK, Shulman NJ, Stergachis AB, Joyner SA, Yan P, Whiteaker JR, Halusa GN, Schilling B, et al. Panorama: a targeted proteomics knowledge base. J Proteome Res. 2014;13:4205–10.
Khudyakov JI, D’Haeseleer P, Borglin SE, Deangelis KM, Woo H, Lindquist EA, Hazen TC, Simmons BA, Thelen MP. Global transcriptome response to ionic liquid by a tropical rain forest soil bacterium, Enterobacter lignolyticus. Proc Natl Acad Sci USA. 2012;109:E2173–82.
Bokinsky G, Baidoo EE, Akella S, Burd H, Weaver D, Alonso-Gutierrez J, Garcia-Martin H, Lee TS, Keasling JD. HipA-triggered growth arrest and beta-lactam tolerance in Escherichia coli are mediated by RelA-dependent ppGpp synthesis. J Bacteriol. 2013;195:3173–82.
Authors’ contributions
Conceptualization of the project: AM TE PD. Strain construction, molecular biology, analytical chemistry: TE RAH PD VB EB. Illumina Sequencing and genome assembly: AL JM RAH. Proteomic analysis: YC CJP. Interpreted results: TE AM PD EB YC CJP. Drafted and revised the manuscript: TE AM LB PD CJP. All authors contributed feedback to the manuscript for publication. All authors read and approved the final manuscript.
Acknowledgements
We thank James Sun (LBNL) and Maren Wehrs (LBNL) for sharing the ILs used in this study. We also thank Steven Singer (LBNL) and Gabriella Papa (LBNL) for technical guidance with the FT-IR. Illumina sequencing was provided by the Department of Energy Joint Genome Institute (JGI, https://jgi.doe.gov/). We thank Christa Pennacchio (JGI) for technical assistance and Leah Freeman Sloan (LBNL) for proof reading the manuscript. This work was part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the U. S. Department of Energy, Office of Science, Biological and Environmental Research, through contract (DE-AC02-05CH11231) between Lawrence Berkeley National Laboratory and the U. S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study, except for the raw Illumina sequencing reads for identification of the cydC mutation, are included in this published article and its additional information files. The raw Illumina sequencing reads are permanently accessible at the Joint Genome Institute/LBL at the following hyperlink: https://perma.cc/PJN4-ZDK7.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Figure S1.
Viability of E. coli DH1 pTRC-cydC strains. E. coli DH1 cells over-expressing Ptrc-cydD-cydC (pTE42) or Ptrc-cydD-cydC-D86G (pTE43) were inoculated with concentrations of the inducer IPTG ranging from 0-32 µM IPTG (“Methods”). Optical density was monitored at 15-minute time points over a 24 hour timeframe. Optical density is plotted on a log10 scale. E. coli DH1 cells overexpressing cydC were slow growing at inducer concentrations from 4-16 µM IPTG and inviable at concentrations above 32 µM IPTG. Cells overexpressing cydC-D86G exhibited less of a decrease of doubling time with IPTG concentrations up to 32 µM IPTG, but were also inviable at 125 µM IPTG. A comparison of doubling time as a function of IPTG concentration for both strains is provided in the bottom panel. Figure S2. E. coli DH1 cydC-D86G Is Also Tolerant to the IL Ethanolamine Acetate (EOA). Wild type and cydC-D86G cells were prepared for growth as in Figure 3 (“Methods”) and grown in the presence or absence of the IL ethanolamine acetate ([EOA]OAc) exactly as described in Figure 1. Both wild-type and cydC-D86G strains had identical growth curves at 30mM and 300mM EOA. Optical density is plotted on a log10 scale. Figure S3. Overexpression of cydAB Does Not Improve Tolerance to [EMIM]OAc. Related to Figure 3. (A, B) E coli BW25113 cells harboring a PLacUV5-cydAB overexpression cassette and a genomic deletion at oppF were prepared for exogenous [EMIM]OAc treatment as described in Figure 3. Cells of the genotype indicated (black circles, WT; red squares, ∆oppF) were tested with or without 50 mM [EMIM]OAc (panel A, with [EMIM]OAc; panel B, without [EMIM]OAc) and with or without 200 µM IPTG (panel A, without IPTG; panel B, with IPTG). Optical density is plotted on a log10 scale. (C) Increased gene dosage of cydC is sufficient to confer tolerance to [EMIM]OAc in E. coli BW25113 strains. Plasmids harboring either cydC (pTE50) or cydC-D86G (pTE88) or an empty vector control (pK18mobsacB) were transformed into E. coli BW25113. Cells of the indicated genotype were then 10-fold serially diluted onto solid LB agar media supplemented with or without 100 mM [EMIM]OAc. LB plates were photographed 1 day after growth at 30 °C; LB 100 mM [EMIM]OAc plates were photographed 4 days after growth at 30 °C. A subtle increase in tolerance against exogenous [EMIM]OAc was observed in strains carrying pTE88 (cydC-D86G) over pTE50 (cydC), but both strains were more resistant compared to the empty vector control. Figure S4. Exogenous [EMIM]OAc Concentration Does Not Change in cydC-D86G Strains. Wild-type E. coli DH1 and E. coli DH1 cydC-D86G strains were grown as described in Figure 4. (A) Measurement of [EMIM]OAc in growth media by FTIR analysis. After ~20 hours of growth, the cell culture media was subjected to room temperature FTIR analysis. Left panel, standard curve using serial dilutions of [EMIM]OAc standard. Right panel, quantification of [EMIM]OAc in biological triplicate samples. No significant difference in [EMIM]OAc concentration was detected between wild-type, cydC-D86G, or growth media alone. (B) Measurement of [EMIM]OAc in growth media by HPLC-MS. Left panel, standard curve using serial dilutions of [EMIM]OAc. Right panel, quantification of [EMIM]OAc by HPLC-LCMS in biological triplicate samples. Figure S5. Specific production of limonene and isopentenol normalized to cell density (biomass). Samples prepared and described in Figure 5 were normalized to cell density, as measured as optical density; Absorbance at 600 nm.
Additional file 2.
Phylogenetic analysis of cydC and cydB genomic co-occurrence in bacterial genomes.
Additional file 3.
Shotgun proteomics raw data for cydC-D86G and control strains.
Additional file 4.
Targeted MRM proteomics raw data for cydC-D86G and control strains.
Additional file 5.
Strains and plasmids used in this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Eng, T., Demling, P., Herbert, R.A. et al. Restoration of biofuel production levels and increased tolerance under ionic liquid stress is enabled by a mutation in the essential Escherichia coli gene cydC. Microb Cell Fact 17, 159 (2018). https://doi.org/10.1186/s12934-018-1006-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-018-1006-8