
Abstract
Background
As regulators of multifunctional metalloproteinases including MMP, ADAM and ADAMTS families, tissue inhibitors of metalloproteinases (TIMPs) play a pivotal role in extracellular matrix remodeling, which is involved in a wide variety of physiological processes. Since abnormal metalloproteinase activities are related to numerous diseases such as arthritis, cancer, atherosclerosis, and neurological disorders, TIMPs and their engineered mutants hold therapeutic potential and thus have been extensively studied. Traditional productions of functional TIMPs and their N-terminal inhibitory domains (N-TIMPs) rely on costly and time-consuming insect and mammalian cell systems, or tedious and inefficient refolding from denatured inclusion bodies. The later process is also associated with heterogeneous products and batch-to-batch variation.
Results
In this study, we developed a simple approach to directly produce high yields of active TIMPs in the periplasmic space of Escherichia coli without refolding. Facilitated by disulfide isomerase (DsbC) co-expression in protease-deficient strain BL21 (DE3), N-TIMP-1/-2 and TIMP-2 which contain multiple disulfide bonds were produced without unwanted truncations. 0.2–1.4 mg purified monomeric TIMPs were typically yielded per liter of culture media. Periplasmically produced TIMPs exhibited expected inhibition potencies towards MMP-1/2/7/14, and were functional in competitive ELISA to elucidate the binding epitopes of MMP specific antibodies. In addition, prepared N-TIMPs were fully active in a cellular context, i.e. regulating cancer cell morphology and migration in 2D and 3D bioassays.
Conclusion
Periplasmic expression in E. coli is an excellent strategy to recombinantly produce active TIMPs and N-TIMPs.
Similar content being viewed by others

Background
Remodeling of the extracellular matrix (ECM) is a critical event in numerous physiological processes (e.g. growth, wound repair, and embryogenesis) [1], and pathological procedures such as arthritis, fibrosis, tumor metastasis, and neurological disorders [2, 3]. Changes in ECM composition are mediated by families of multi-domain extracellular proteinases including MMPs (matrix metalloproteinases), ADAMs (a disintegrin and metalloproteinases), and ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs). The enzymatic activity and zymogen activation of these metalloproteinases are governed by a group of endogenous proteins named tissue inhibitors of metalloproteinases (TIMPs) [4]. Four human TIMPs have been identified (TIMP-1 to -4, sharing 40% sequence identity), among which TIMP-1/-3 are glycoproteins, whereas TIMP-2/-4 are not glycosylated [5]. All mammalian TIMPs have 12 conserved cysteine residues forming six disulfide bonds that are essential for their functions and structural integrity [1]. N-terminal domains of TIMPs (~125 aa, N-TIMPs) and their C-terminal domains (~65 aa, C-TIMPs) have three disulfide bonds each. N-TIMPs alone fold to stable and native structures carrying full inhibition activities to MMPs and other disintegrin-metalloproteinases [6,7,8]. C-TIMPs are more divergent, and able to improve inhibition selectivities and binding efficiencies [9]. Investigations with isolated MMP domains and their crystal structures have indicated that N-TIMPs interact directly with the catalytic domain of MMPs to form a stable inactive complex [10].
TIMPs inhibit MMPs with various inhibition constants (K I s) ranging from low to subnanomolar, implicating that they have distinct functions in vivo [1]. In addition to MMPs, several members of the ADAM and ADAMTS families are regulated by TIMPs as well. Among TIMPs, TIMP-3 exhibits the broadest inhibition spectrum. To improve their inhibition potency and selectivity among individual MMPs, N-TIMPs have been engineered by directed evolution using phage display [11] and TIMP chimeras have been designed to expand specificity [12]. As a consequence of their metalloproteinase inhibition activity, TIMPs exhibit biological functions such as promoting cell proliferation, inhibiting angiogenesis, inhibiting transendothelial migration and regulating migration [13,14,15,16,17,18]. Because of these important physiological processes TIMPs regulate in vivo and in vitro, TIMPs have been the focus of many biochemistry and cell biology researches, and thus steady supplies of active human TIMPs at milligram scales are vital.
Mammalian and insect cells have been employed for producing properly folded recombinant TIMPs [6, 7, 9, 19,20,21]. With merits of high yield and low cost, bacterial expression systems have also been attempted [21–23]. However, overexpression of (N-)TIMPs in Escherichia coli, even when fused with highly soluble MBP (maltose-binding protein), led to improper or incomplete folding or the formation of insoluble inclusion bodies. To obtain their native conformation, the insoluble TIMPs require refolding via manifold denaturation–renaturation steps, i.e. urea solubilization and gradient dialysis. This labor-substantial and time-consuming preparation of TIMPs (usually 3–5 days) significantly impedes studies of TIMPs, especially when TIMP variants are needed [24, 25]. Although successful refolding of TIMPs has been reported in literature [8, 21, 22, 24,25,26,27,28,29], due to the various chemical–physical properties and characteristics of TIMPs, the optimal procedures must be tailored for each TIMP. In addition to deficient yields, other inevitable limitations often include poor reproducibility, heterogeneous oligomeric products, and difficulty of scale-up [22, 26].
Encouraged by functional productions of a variety of recombinant human proteins containing multiple disulfide bonds, i.e. T cell receptors, antibodies and tPA (tissue plasminogen activator) [30,31,32], in the oxidative periplasm of E. coli, we hypothesize that periplasmic expression will result in soluble and active TIMPs. In addition to providing an oxidative environment that promotes the formation of disulfide bonds of (N-)TIMPs, periplasmic expression can enhance the correct protein folding due to multiple molecule chaperons (e.g. SurA, PpiA, Ppid, FkpA, and Skp) [33] and a slow processing rate controlled by secretion machineries [34]. Our previous study directly expressed functional MMP-14 catalytic domain (cdMMP-14) in periplasmic space of E. coli [31], and we aim to apply a similar approach for active TIMP production. In this study, by optimizing co-expression of a set of disulfide bond enzymes (Dsb proteins) and selecting a proper expression host, soluble and monomeric (N-)TIMPs were produced in periplasm with high yields (Fig. 1). Periplasmically produced (N-)TIMPs exhibited their biological activities in MMP inhibition assays and cell migration tests. We expect the novel method described here—direct production of functional (N-)TIMPs in E. coli without refolding—could greatly expedite many facets of in vitro and in vivo studies associated with metalloproteinases and ECM remodeling.

Direct production of soluble (N-)TIMPs in E. coli periplasm and their biochemical and cellular function characterizations. Unfolded TIMPs with free cysteines were expressed in cytoplasm and secreted to periplasmic space, where periplasmic chaperones, especially DsbC (a disulfide isomerase), resolved incorrect disulfide bonds, resulting in properly folded TIMPs. Following enzymatic and osmotic treatments, high yields of soluble (N-)TIMPs were purified from periplasmic preparation. The purified (N-)TIMPs were subjected to function tests both biochemically and in the cellular context. GPC gel permeation chromatography
Results
Production of soluble TIMPs in E. coli periplasm with high yields
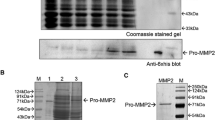
Full-length TIMP-1/-2/-3/-4 and N-terminal domains of TIMP-1/-2/-3 were constructed at the downstream of a Plac promoter and a pelB leader peptide sequence. Crystallography of MMP-TIMP complexes suggested that N-terminal residues CXCX of TIMPs directly interact with MMP reaction cleft [35], and TIMP-2 variant with an alanine appended to the amino terminus (Ala+TIMP-2) was inactive [28]. Therefore, a hexa-histidine tag was genetically tagged to the C-termini of (N-)TIMPs for detection and affinity purification. TIMP constructs were transformed to E. coli Jude-I for expression. Initial tests indicated that no induction resulted in a higher soluble expression than induction with 1 µM IPTG, a similar phenomenon observed for cdMMP-14 expression [31]. After purification, reducing SDS-PAGE (Fig. 2a) showed single and strong bands of N-TIMP-1/2 (15 kDa) and TIMP-2 (23 kDa), consistent with their calculated MWs. Particularly, 0.5 and 1.4 mg of purified N-TIMP-1/-2 were yielded per liter of culture media. However, TIMP-1/-4 were expressed at much lower levels. Purified TIMP-1 sample showed two bands, one for mature TIMP-1 (22 kDa), and the other band likely associated with unprocessed TIMP-1 having the pelB leader signal peptide (27 kDa). In the case of TIMP-4, unwanted truncation was detected at 17 kDa, in addition to the full-length TIMP-4 at 23 kDa, and bands corresponding to N-TIMP-3 and TIMP-3 were not present in their purified samples (Fig. 2a).

Periplasmic production of (N-)TIMPs and expression condition optimization. a Reducing SDS-PAGE of purified (N-)TIMPs stained with Coomassie blue. Red arrows indicate the target bands. b Effect of periplasmic folding modulators (DsbA and/or DsbC) on expression efficiencies of (N-)TIMPs analyzed by Western blotting using anti-6×His antibody. 1P indicates DsbA and DsbC were under one P BAD promoter and 2P represents DsbA and DsbC were under two separated P BAD promoters. Red arrows indicate the target bands. c Effect of BL21(DE3) on reducing unwanted truncations analyzed by Western blotting. Same amounts of cells were used in b and c
Previous studies have demonstrated that periplasmic folding modulators can improve the soluble expression and homogeneity of proteins containing disulfide bonds [32, 36,37,38]. To increase expression levels of soluble TIMP-1/-3/-4 and N-TIMP-3, we chose DsbA and DsbC as folding modulators to catalyze the formation and isomerization of disulfide bridges, respectively [33]. To investigate the optimal effect of DsbA and/or DsbC, Jude-I cells carrying the TIMP genes were co-transformed with one of four chaperone plasmids: pBAD-DsbA, pBAD-DsbC, pBAD-AC1P (both DsbA and DsbC under one P BAD promoter), and pBAD-AC2P (both DsbA and DsbC under two separate P BAD promoters). After cultured in 2×YT media overnight at 30 °C without IPTG, crude periplasmic fractions were prepared for analysis by Western blotting using anti-6×His-HRP. As results shown in Fig. 2b, DsbC (either alone or with DsbA in the 2P format) dramatically raised the yields of TIMP-1/-3 and N-TIMP-3, while DsbA alone did not exhibit significant improvements of TIMP expression. The result that DsbC (disulfide bonds isomerase) was far more efficient than DsbA (a thiol disulfide oxidoreductase) for TIMPs production suggests that presumably resolving the non-native disulfide bonds was the key for proper folding of TIMPs. The suboptimal result of co-expression of both DsbA/C in 1P format was probably due to the inadequate expression level of DsbC that was at downstream of the polycistronic construct. In addition, DsbC alone also significantly enhanced TIMP-4 production at a similar degree as TIMP-1/-3 (Fig. 2b).
Although the amounts of expression were significantly improved by DsbC, unacceptable levels of truncated species were still present in (N-)TIMPs samples (Fig. 2b). We hypothesized that these truncations were digestion products by E. coli endogenous proteases, and thus using protease-deficient strain, e.g. BL21 (DE3) could solve the problem. Western blotting analysis confirmed that when BL21 (DE3) was used with DsbC co-expression, truncations were dramatically reduced and only single and strong bands of TIMP-1/-3/-4 were shown (Fig. 2c). With facilitation of DsbC, soluble TIMP-1/-2 were produced in periplasmic fractions of BL21 (DE3) with yields of 0.2 mg purified proteins per liter of 2×YT media.
The quality of purified TIMPs was characterized by gel permeation chromatography (GPC). N-TIMP-2 sample displayed a single peak at ~15 kDa associated with its monomer. No aggregates or oligomers were detected, suggesting high quality of the preparation (Fig. 3a). GPC analysis of prepared N-TIMP-1 showed a major peak (~15 kDa) corresponding to its monomer, with presence of trace amounts of oligomers (Fig. 3b). However, compared with N-TIMP-1 sample prepared without DsbC co-expression (Additional file 1: Figure S1), the degree of oligomerization was dramatically reduced, suggesting formation accurate disulfide bonds is critical for TIMPs production.

Size exclusion chromatography of purified a N-TIMP-2 and b N-TIMP-1. The size-exclusion column was equilibrated with 50 mM HEPES (pH 7.5) and 150 mM NaCl. 100 µL of 500 µg/mL N-TIMP-1/-2 was loaded to a superdex™ 75 10/300 GL column (10 mm × 300 mm) at a flow rate of 0.5 mL/min. Chromatograms were obtained by monitoring absorbance at 280 nm. The molecular mass of N-TIMP-1/-2 was estimated by its retention time and comparison with these of standard molecular mass markers, e.g. ovalbumin (43 kDa) and lysozyme (14.3 kDa)
Produced TIMPs exhibited expected inhibition potencies towards a panel of MMPs
Because inhibitions of TIMPs to metalloproteinases are not highly specific, four MMPs were chosen to test the functions of purified (N-)TIMP-1/-2: MMP-1 (collagenase 1), MMP-2 (gelatinase A), MMP-7 (matrilysin), and cdMMP-14 (membrane type 1-MMP). Using quenched fluorescent peptide substrates, MMPs’ enzymatic activities were measured in the presence of 0.98 nM–1 µM purified TIMPs. As results shown in Fig. 4, TIMP-2 and N-TIMP-1/-2 efficiently inhibited MMP-1/-2/-7 cleaving the peptide substrates in a dose-dependent manner with IC50 values ranging from 3 to 72 nM. However, N-TIMP-1 was a relatively weak inhibitor to cdMMP-14, an observation consistent with literature [5, 27].

Inhibitory activities of (N-)TIMPs toward MMPs. Semi-quantitative IC50s of N-TIMP-1 were determined as 46, 65, and 57 nM for MMP-1/2/7 respectively; IC50s of N-TIMP-2 were 3, 6, 38, and 72 nM for MMP-1/2/7 and cdMMP-14 respectively; and IC50s of TIMP-2 were 11, 2, 22, and 22 nM for MMP-1/2/7 and cdMMP-14 respectively. Error bars represent standard deviations from two independent measurements
Inhibition constants (K I s) were further calculated for direct comparison with the TIMPs produced through traditional approaches (i.e. mammalian cells or refolding from inclusion body). Results (Table 1) indicated that, except N-TIMP-1 as a weak inhibitor to cdMMP-14, all measured K I s were in nanomolar to subnanomolar scale as expected. Particularly, N-TIMP-1 inhibited MMP-1/-2/-7 with K I s of 7.21 ± 0.33, 10.98 ± 3.76, and 4.62 ± 0.44 nM, respectively. N-TIMP-1 behaves with a similar potency to MMP-7 as reported [27], but its K I s to MMP-1/-2 were 2–27-folds less potent than literature values depending on the studies [21, 24, 27], suggesting the possibility that only portion of prepared N-TIMP-1 was active. However, for periplasmically produced N-TIMP-2, its calculated K I s (0.65 ± 0.14, 0.80 ± 0.03, 2.86 ± 0.94, and 3.96 ± 0.53 nM for MMP-1/-2/-7/-14cd, respectively) well matched with literature data [25, 27], indicating high quality of N-TIMP-2 preparation. The inhibition potencies of TIMP-2 were measured as 2.51 ± 0.01, 0.39 ± 0.09, 2.74 ± 0.09, and 0.34 ± 0.04 nM for MMP-1/-2/-7/-14cd, respectively. Among tested MMPs, only MMP-2 has TIMP-2 inhibition data available in literature, i.e. K I = 0.6 ± 0.3 nM [28], which well agreed with our results (Table 1). Collectively, both N-TIMP-2 and TIMP-2 exhibited expected K I s, suggesting their correct folding in periplasm. Interestingly, compared with N-TIMP-2, TIMP-2 displayed a significantly higher potency to cdMMP-14, implying that the C-terminal domain of TIMP-2 may contribute recognition of MMPs as well [39].
N-TIMP-2 was functional in antibody epitope characterization
With structural and inhibition mechanism information available for MMP–TIMP complexes [10, 35, 39], TIMPs have been employed to provide insights on epitopes and affinities of monoclonal antibodies that bind to (and inhibit) MMPs [40]. To investigate whether periplasmically produced TIMPs are capable in such functional assays, competitive ELISAs were conducted in which fixed concentration of Fabs of interest was mixed with increased amounts of TIMP and incubated in wells coated with the associated MMP, and captured Fabs were detected by anti-Fab-HRP for signal development. In our previous studies, a panel of Fabs inhibiting MMP-14 has been isolated from phage display antibody libraries [41]. Three Fabs—3A2 (inhibitory, K I = 9.4 ± 1.4 nM), 3D9 (inhibitory, K I = 27 ± 3.5 nM), and 3E9 (non-inhibitory)—were therefore used to test N-TIMP-2 functionality in this study. Results indicated that N-TIMP-2 directly competed with Fabs 3A2 and 3D9 on binding to cdMMP-14 with sigmoidal dose–response curves (Fig. 5). As N-TIMP-2 concentration increased, lower amounts of Fabs 3A2 or 3D9 bound to cdMMP-14. Between these two Fabs, 3A2 responded at a higher N-TIMP-2 concentration, consistent with our measurements that 3A2 is more potent than 3D9 [41]. In contrast, binding of Fab 3E9 to cdMMP-14 did not significantly respond to N-TIMP-2 concentration. These results agreed with our previous observations that Fabs 3A2 and 3D9 bound to MMP-14 reaction cleft vicinity therefore directly competed with N-TIMP-2, while Fab 3E9 was non-inhibitory antibody likely having an epitope not overlapped with the binding site of N-TIMP-2 [41]. Overall, periplasmically produced N-TIMP-2 was successfully used for antibody characterizations by competitive ELISA, an effective method to identify and study antibody inhibitors.

Competitive ELISA between N-TIMP-2 and Fabs for cdMMP-14. Fixed concentrations of Fabs (10 nM for 3A2 and 3D9, and 20 nM for 3E9) were incubated with increasing concentration of N-TIMP-2 on ELISA plates coated with cdMMP-14. The captured Fabs were detected by anti-Fab-HRP for signal development
N-TIMP-1/-2 decreased directionality, and N-TIMP-2 altered migration speed of tumor cells
To test the role of TIMPs in regulating migration, we treated human breast adenocarcinoma MDA-MB-231 cells with 50 nM N-TIMP-1 or -2 and assessed directed cell migration in response to aligned collagen fibers (contact guidance) in 2D and 3D environments. This final concentration was chosen so that MMPs like MMP-2 would be inhibited with either N-TIMP-1 or -2, whereas MMPs like MMP-14 would primarily be inhibited with N-TIMP-2. The cells on 2D contact guidance cues were grouped in four different morphological shapes as shown in Fig. 6A–D. These categories included cells that were spindle shaped (I), branched (Y), bent (Γ) or spread (O). Most cells are identified as spindle shaped, however the addition of N-TIMP-1 or -2 decreased the fraction that were spindle shaped and increased branched, bent and spread, although not significantly. Furthermore, N-TIMP-2 was more effective in forcing this change. The cells suspended in 3D contact guidance cues were grouped in two morphological shapes: spindle shaped (I) or round (O) (Fig. 6E, F). The addition of the N-TIMP-1 or -2 did not have a statistically significant effect on the overall fractions in each category.

Analysis of N-TIMPs on MDA-MB-231 cell behavior in 2D and 3D contact guidance environments. Cells on 2D substrates (A–D) or in 3D environments (E–F) where grouped based on shape and the fraction of cells in each shape were calculated: A 2D spindle, B 2D branched, C 2D bent, D 2D spread, E 3D spindle and F 3D round. Calibration bar is 50 μm. G Fraction of cells that were of different shapes (N cells > 79, N experiments > 3). Error bars are 95% confidence intervals calculated using Matlab. H Aspect ratio, I migration speed and J directionality in both 2D (black) and 3D (grey) environments (N cells > 79, N experiments > 3). Error bars are 95% confidence intervals calculated using Matlab. Statistical significance was tested using a t test with p < 0.05 and indicated with bars
While N-TIMP-1 and -2 only marginally affected the cell morphology shape in 2D and 3D contact guidance systems, they significantly affect other attributes. N-TIMP-2 increased the aspect ratio, cell length divided by cell width, during 2D contact guidance, whereas N-TIMP-1 decreases the aspect ratio during 3D contact guidance (Fig. 6H). Cell migration speed responded similarly. While N-TIMP-2 increased speed on 2D contact guidance cues, it decreased speed on 3D contact guidance cues (Fig. 6I). On the other hand, cell directionality changed in the same direction across the 2D and 3D systems. N-TIMP-2 decreases directional fidelity in both 2D and 3D contact guidance environments with the most robust response seen during 3D contact guidance (Fig. 6J). N-TIMP-1, on the other hand, had no effect on directional fidelity in 2D, but decreased directional fidelity in 3D contact guidance system, albeit to a lesser extent than N-TIMP-2.
Discussion
TIMPs expressed in connective tissues play pivotal roles in extracellular matrix metabolism by inhibiting MMP activities in vivo [2, 3]. Indeed, tumor cell invasion and metastasis can be impeded by up-regulation of TIMPs expression or by an exogenous supply of TIMPs [42,43,44]. Therefore, a constant and reliable supply of active TIMPs is important for fundamental research and therapeutic development targeting unbalanced metalloproteinases.
Recombinant production of active human TIMPs and their N-terminal domains, usually employs eukaryotic expression systems, chiefly due to disulfide bond formation and glycosylation [6, 7, 9, 21]. Specifically, TIMP-1 has two N-linked glycosylation sites at Asn30 and Asn78. Although aglycosylated TIMP-1 shows uncompromised inhibition potency, it has been suggested that N-glycosylation involves in its folding, solubility and transport to cell surface [6, 7, 45,46,47]. The glycosylation status of TIMPs has been largely disregarded in clinical samples from patients with cancer, but their role in cancer progression may confound expectations [48]. Glycosylated TIMP-1, but not aglycosylated one, exerts influence of tumor formation and growth in the early phase [49]. Recently, production of N-glycosylated proteins in E. coli was achieved by utilizing and engineering the N-linked glycosylation system of Campylobacter jejuni [50]. With current endeavors to further develop this approach for complicated glycosylations [51], our system would benefit from all these advances for production of fully glycosylated TIMP-1/-3 in E. coli.
Numerous studies have been conducted to optimize the refolding process, especially to focus on restoring the multiple disulfide bridges [8, 21, 22, 24,25,26,27,28,29]. Nevertheless, generating biologically active TIMPs from E. coli inclusion bodies is considerably challenging, because TIMPs contain 12 cysteine residues, all of which participate in intramolecular disulfide linkages. The aim of present study was to develop an expression system without tiresome refolding process to produce active human TIMPs at high yields that are sufficient for biochemical and cellular experiments. In distinct contrast to previous studies, we employed periplasmic space of E. coli to achieve the purpose. By optimizing periplasmic folding modulators in conjunction with expression host and conditions, full length TIMPs and their N-terminal domains were solubly produced at yields of 0.2–1.4 mg purified proteins per liter of culture (Fig. 2). Particularly, periplasmic co-expression of a thiol:disulfide interchange protein DsbC dramatically increased the titers of TIMPs, presumably due to its disulfide isomerization activity by correcting the non-native disulfide bonds, as well as to its general chaperone activity to prevent TIMPs aggregation. Impressively, purified N-TIMP-2 showed as a clean single peak associated with monomer in gel permeation chromatography, without aggregates or oligomeric species presented (Fig. 3a).
The activities and functionalities of periplasmically prepared TIMPs were then confirmed by a set of experiments: (1) inhibition assays against a panel of MMPs, including direct comparison of TIMP-2 and N-TIMP-2 inhibition potencies for the first time (Fig. 4; Table 1); (2) characterization of MMP specific antibodies (Fig. 5); and (3) cell morphology and migration assays in 2D and 3D extracellular matrix (Fig. 6).
To our best knowledge, it is the first study to validate direct production of active human TIMPs in periplasm of E. coli. The approach without refolding is straightforward and reliable with less lot-to-lot variation. And high yields and homogeneity of the products indicate that our method is suitable for designed or randomized mutagenesis studies to investigate the biological roles of TIMPs and lead for therapeutic development of engineered TIMP variants. Furthermore, the periplasmic preparation is simple and fast, i.e. the enzymatic and osmotic treatment followed by centrifugation can be finished in ~40 min. In this aspect, we expect that the periplasmic preparation of TIMPs can greatly facilitate downstream processing and make this approach economically attractive.
Conclusions
Direct production of high yields of active (N-)TIMPs without refolding was achieved in the periplasmic space of protease-deficient E. coli strain by co-expressing disulfide isomerase (DsbC). Periplasmically produced TIMPs exhibited expected inhibition potencies towards a panel of MMPs, and functional in competitive ELISA and in a cellular context, i.e. regulating cancer cell morphology and directed migration in 2D and 3D bioassays.
Methods
Materials
Restriction enzymes and Vent DNA polymerase were obtained from NEB. Human MMPs-1/2/7 were purchased from AnaSpec. The catalytic domain of human MMP-14 (cdMMP-14) was produced and biotinylated as previously described [31]. The following vectors and cell lines were from lab stock: periplasmic expression vector pMopac16 [52], pBAD33 [53], DsbA/C chaperon plasmids [54] pBAD-DsbA, pBAD-DsbC, pBAD-AC1P (both DsbA and DsbC under one P BAD promoter) and pBAD-AC2P (DsbA and DsbC under two separated P BAD promoters), and E. coli strains Jude-I [DH10B F’::Tn10(Tetr)] and BL21(DE3). A human mammary basal/claudin low carcinoma cell line (MDA-MB- 231, ATCC) was cultured in Dulbecco’s Modified Eagles Medium (DMEM) (Sigma-Aldrich) containing 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin–streptomycin (pen-strep) (Gibco) at 37 °C in 5% CO2. Imaging media for MDA-MB-231 cells was the same as the subculturing media, with the exception that no phenol red was included and that 12 mM HEPES (Sigma-Aldrich) was included.
Cloning and expression of (N-)TIMPs
The DNA fragments encoding human TIMP-1/2/3/4 were synthesized (IDT) and cloned into pMopac16, which carries a lac promoter and a pelB leader. The genes of N-TIMP-1/-2/-3 were PCR amplified (residues 1–126, 1–127 and 1–121 respectively) and cloned into pMopac16. Transformed E. coli Jude-I cells were grown in 2×YT media supplemented with 35 µg/mL chloramphenicol (2×YT/Chlor) for (N-)TIMPs expression overnight at 30 °C without IPTG. For co-expression with periplasmic folding modulators, the chloramphenicol acetyltransferase genes on the chaperone plasmids were replaced with β-lactamase genes. Double transformed Jude-I or BL21(DE3) containing pBAD-DsbA/-DsbC/-AC1P/-AC2P and pMopac16-(N-)TIMPs were cultured at 30 °C overnight in 2×YT media supplemented with 100 µg/mL ampicillin, 35 µg/mL chloramphenicol and 0.2% arabinose (2×YT/Amp/Chlor/arabinose) without IPTG.
Periplasmic fractions preparation and recombinant (N-)TIMPs purification
Periplasmic fractions were prepared as previously reported [31]. Briefly, three OD cells were harvested and resuspended in 100 μL 200 mM Tris–HCl pH 7.5, 20% sucrose, 30 U/μL lysozyme, and then incubated for 10 min at room temperature. The mixture was then treated by osmotic shock with 100 μL ice-cold ddH2O followed by incubation on ice for 10 min. The mixture was centrifuged at 15,000×g for 2 min, and the recovered supernatant was analyzed by SDS-PAGE and Western blotting with anti-6×His-HRP (Abcam) and chemiluminescent substrate (Thermo Scientific). The (N-)TIMPs were purified from periplasmic preparations by Ni–NTA agarose beads following manufacturer’s protocols (Qiagen). The homogeneity of purified proteins was confirmed by SDS-PAGE. Overnight dialysis was conducted to remove residual imidazole, and the concentrations of purified (N-)TIMPs were determined by NanoDrop (Thermo), before storage in 20% glycerol at −80 °C.
Gel permeation chromatography
GPC analyses were performed on an ÄKTAprime using a superdex 75 10/300 GL size-exclusion column (GE Healthcare) equilibrated with 50 mM HEPES pH 7.5, 150 mM NaCl. 100 µL 500 µg/mL N-TIMP-2 was injected at a flow rate of 0.5 mL/min to obtain chromatograms of absorbance at 280 nm. The molecular mass of N-TIMP-2 was estimated based on the retention times of standard proteins including ovalbumin (43 kDa) and lysozyme (14.3 kDa) (Sigma-Aldrich).
FRET inhibition assay
Concentrations of active MMPs were titrated by GM6001 (a highly potent broad-spectrum MMP inhibitor, EMD). In 96-well black assay plates (Corning), 60/60/250/500 nM MMP-1/-2/-7/-14cd was incubated with 0.98–1000 nM (N-)TIMPs for 30 min at room temperature, and 1 μM M-2350 substrate (Bachem) was added to start the reactions. The fluorescent signals with excitation at 328 nm and emission at 393 nm were monitored using a Synergy H4 microplate reader (BioTek). Initial velocities were calculated to determine IC50, and inhibition constants (K I s) were obtained using the following equation for slow tight-binding inhibitors [5]:
[I] t is the total inhibitor concentration, [E] t is the total enzyme concentration. v 0 is the rate of substrate hydrolysis in the absence of TIMP (M · s−1), v s is the equilibrium rate of substrate hydrolysis reached after inhibition (rate of hydrolysis in the inhibited steady state) (M · s−1) for each TIMP concentration, [I] t .
Competitive ELISA
12 μM N-TIMP-2 was two-fold serially diluted and incubated with Fabs of interest (10 nM 3A2, 10 nM 3D9, or 20 nM 3E9) for 15 min in streptavidin microplate wells coated with 5 μg/mL biotinylated cdMMP-14. After washing, bound Fabs were detected by anti-Fab-HRP and TMB solution, then stopped with 1 M H2SO4. OD450 signals were measured using a microplate reader.
Imaging at 2D environment
For 2D cell imaging, a 15 × 15 mm piece of muscovite mica (highest grade VI, Ted Pella, Redding, CA, USA) was freshly cleaved using tape. High concentration non-pepsin treated rat tail collagen type I (Corning, Corning, NY, USA) was diluted to 10 μg/mL in 50 mM Tris–HCl pH 9.2 and 200 mM KCl, and incubated on the mica at room temperature for 3–6 h [55]. After incubation, the mica was washed with distilled water, laid against the edge of a tissue culture dish then allowed to dry overnight. Collagen substrates were seeded with MDA-MB-231 cells (~6000 cells/cm2), which were treated with 50 nM N-TIMP-1 or N-TIMP-2. After 2 h of incubation, substrates with cells attached to the fibrils were inverted onto two strips of double-sided tape attached to a microscope slide to generate a flow chamber. The chamber was filled with imaging media, sealed and imaged by phase contrast microscopy on a heated stage at 37 °C every 2 min for 12 h.
Cell imaging in 3D gels
For 3D imaging, the cells were trypsinized and suspended at a concentration of 600,000 cells/mL in a 2 mg/mL non-pepsin-treated rat collagen type I solution. 50 nM N-TIMP-1 and N-TIMP-2 were added to this solution prior to the addition of the collagen. A 125 μL volume of the solution was pipetted into a MatTek dish (MatTek Corporation, Ashland, MA, USA) and allowed to polymerize at room temperature for 45 min. After polymerization, 125 μL of imaging media was added to the top of the gel. To seal the chamber, a 5-mm thick pad of polydimethylsiloxane (PDMS) (Dow) punctured with an acupuncture needle was placed on top and sealed with food grade lubricant (McGlaughlin Oil Company, Columbus, OH). The acupuncture needle was then rotated one full rotation to align the collagen fibers and placed in a 37 °C incubator. After 24 h, the chambers were removed from the incubator and imaged every 2 min for 8 h. The images were analyzed using the MTrackJ plugin in ImageJ which allows the user to track the cells on an image-by-image basis. The plugin generates the x–y coordinates of the cell at each time point. Trajectories were analyzed using a custom Matlab script to calculate a migration speed. Directionality was determined using another custom Matlab script. The angle between the direction of the migration and a radial vector field originating from the needle was used to calculate directionality with the following equation:
Aspect ratio was calculated by measuring the length of a cell and dividing it by the width of that same cell. Analysis of cell morphology was completed by binning cell shape based on the shape created by the extensions after the cells had time to adhere to its environment: 9 h for 2D and 24 h for 3D. 95% confidence intervals were calculated using Matlab with a sample of at least 79 cells. A two tailed t test at p ≥ 0.05 was conducted to identify statistical differences between the N-TIMP conditions and that of the control.
Abbreviations
- TIMP:
-
tissue inhibitor of metalloproteinase
- MMP:
-
matrix metalloproteinase
- ECM:
-
extracellular matrix
- ADAM:
-
a disintegrin and metalloproteinase
- ADAMTS:
-
a disintegrin and metalloproteinase with thrombospondin motifs
- DsbA and DsbC:
-
disulfide oxidoreductases A and C
References
Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta. 2000;1477:267–83.
Reynolds JJ, Meikle MC. Mechanisms of connective tissue matrix destruction in periodontitis. Periodontol 2000. 1997;14:144–57.
Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci. 1998;21:75–80.
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803:55–71.
Troeberg L, Nagase H. Analysis of TIMP expression and activity. Methods Mol Med. 2007;135:251–67.
Murphy G, Houbrechts A, Cockett MI, Williamson RA, O’Shea M, Docherty AJP. The N-terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry. 1991;30:8097–102.
Williamson RA, Bartels H, Murphy G, Freedman RB. Folding and stability of the active N-terminal domain of tissue inhibitor of metalloproteinases-1 and -2. Protein Eng. 1994;7:1035–40.
Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J Biol Chem. 2001;276:12501–4.
Willenbrock F, Crabbe T, Slocombe PM, Sutton CW, Docherty AJP, Cockett MI, O’Shea M, Brocklehurst K, Phillips IR, Murphy G. The activity of the tissue inhibitors of metalloproteinases is regulated by C-terminal domain interactions: a kinetic analysis of the inhibition of gelatinase A. Biochemistry. 1993;32:4330–7.
Fernandez-Catalan C, Bode W, Huber R, Turk D, Calvete JJ, Lichte A, Tschesche H, Maskos K. Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J. 1998;17:5238–48.
Bahudhanapati H, Zhang Y, Sidhu SS, Brew K. Phage display of tissue inhibitor of metalloproteinases-2 (TIMP-2): identification of selective inhibitors of collagenase-1 (metalloproteinase 1 (MMP-1)). J Biol Chem. 2011;286:31761–70.
Duan JX, Rapti M, Tsigkou A, Lee MH. Expanding the activity of tissue inhibitors of metalloproteinase (TIMP)-1 against surface-anchored metalloproteinases by the replacement of its C-terminal domain: implications for anti-cancer effects. PLoS ONE. 2015;10:e0136384.
Hayakawa T, Yamashita K, Tanzawa K, Uchijima E, Iwata K. Growth-promoting activity of tissue inhibitor of metalloproteinases-1 (TIMP-1) for a wide range of cells A possible new growth factor in serum. FEBS Lett. 1992;298:29–32.
Stetler-Stevenson WG, Bersch N, Golde DW. Tissue inhibitor of metalloproteinase-2 (TIMP-2) has erythroid-potentiating activity. FEBS Lett. 1992;296:231–4.
Yang TT, Hawkes SP. Role of the 21-kDa protein TIMP-3 in oncogenic transformation of cultured chicken embryo fibroblasts. Proc Natl Acad Sci USA. 1992;89:10676–80.
Shen QA, Lee ES, Pitts RL, Wu MH, Yuan SY. Tissue inhibitor of metalloproteinase-2 regulates matrix metalloproteinase-2-mediated endothelial barrier dysfunction and breast cancer cell transmigration through lung microvascular endothelial cells. Mol Cancer Res. 2010;8:939–51.
Strongin AY. Proteolytic and non-proteolytic roles of membrane type-1 matrix metalloproteinase in malignancy. Biochim Biophys Acta. 2010;1803:133–41.
Clarke MR, Imhoff FM, Baird SK. Mesenchymal stem cells inhibit breast cancer cell migration and invasion through secretion of tissue inhibitor of metalloproteinase-1 and-2. Mol Carcinog. 2015;54:1214–9.
Gomez DE, Lindsay CK, Cottam DW, Nason AM, Thorgeirsson UP. Expression and characterization of human tissue inhibitor of metalloproteinases-1 in a baculovirus-insect cell system. Biochem Biophys Res Commun. 1994;203:237–43.
Liu YE, Wang M, Greene J, Su J, Ullrich S, Li H, Sheng S, Alexander P, Sang QA, Shi YE. Preparation and characterization of recombinant tissue inhibitor of metalloproteinase 4 (TIMP-4). J Biol Chem. 1997;272:20479–83.
Huang W, Suzuki K, Nagase H, Arumugam S, Van Doren SR, Brew K. Folding and characterization of the amino-terminal domain of human tissue inhibitor of metalloproteinases-1 (TIMP-1) expressed at high yield in E. coli. FEBS Lett. 1996;384:155–61.
Cocuzzi ET, Walther SE, Rajan S, Denhardt DT. Expression and purification of mouse TIMP-1 from E. coli. FEBS Lett. 1992;307:375–8.
Nallamsetty S, Waugh DS. A generic protocol for the expression and purification of recombinant proteins in Escherichia coli using a combinatorial His6-maltose binding protein fusion tag. Nat Protoc. 2007;2:383–91.
Huang W, Meng Q, Suzuki K, Nagase H, Brew K. Mutational study of the amino-terminal domain of human tissue inhibitor of metalloproteinases 1 (TIMP-1) locates an inhibitory region for matrix metalloproteinases. J Biol Chem. 1997;272:22086–91.
Butler GS, Hutton M, Wattam BA, Williamson RA, Knäuper V, Willenbrock F, Murphy G. The specificity of TIMP-2 for matrix metalloproteinases can be modified by single amino acid mutations. J Biol Chem. 1999;274:20391–6.
Kleine T, Bartsch S, Bläeser J, Schnierer S, Triebel S, Valentin M, Gote T, Tschesche H. Preparation of active recombinant TIMP-1 from Escherichia coli inclusion bodies and complex formation with the recombinant catalytic domain of PMNL-collagenase. Biochemistry. 1993;32:14125–31.
Hamze AB, Wei S, Bahudhanapati H, Kota S, Acharya KR, Brew K. Constraining specificity in the N-domain of tissue inhibitor of metalloproteinases-1; gelatinase-selective inhibitors. Protein Sci. 2007;16:1905–13.
Wingfield PT, Sax JK, Stahl SJ, Kaufman J, Palmer I, Chung V, Corcoran ML, Kleiner DE, Stetler-Stevenson WG. Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli. Comparison of wild type and amino-terminal alanine appended variant with implications for the mechanism of TIMP functions. J Biol Chem. 1999;274:21362–8.
Williamson RA, Natalia D, Gee CK, Murphy G, Carr MD, Freedman RB. Chemically and conformationally authentic active domain of human tissue inhibitor of metalloproteinases-2 refolded from bacterial inclusion bodies. Eur J Biochem. 1996;241:476–83.
Ward ES. Secretion of T cell receptor fragments from recombinant Escherichia coli cells. J Mol Biol. 1992;224:885–90.
Nam DH, Ge X. Direct production of functional matrix metalloproteinase-14 without refolding or activation and its application for in vitro inhibition assays. Biotechnol Bioeng. 2016;113:717–23.
Qiu J, Swartz JR, Georgiou G. Expression of active human tissue-type plasminogen activator in Escherichia coli. Appl Environ Microbiol. 1998;64:4891–6.
Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22:1399–408.
Miot M, Betton JM. Protein quality control in the bacterial periplasm. Microb Cell Fact. 2004;3:4.
Gomis-Rüth FX, Maskos K, Betz M, Bergner A, Huber R, Suzuki K, Yoshida N, Nagase H, Brew K, Bourenkov GP, Bartunik H, Bode W. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature. 1997;389:77–81.
Schlapschy M, Grimm S, Skerra A. A system for concomitant overexpression of four periplasmic folding catalysts to improve secretory protein production in Escherichia coli. Protein Eng Des Sel. 2006;19:385–90.
Maskos K, Huber-Wunderlich M, Glockshuber R. DsbA and DsbC-catalyzed oxidative folding of proteins with complex disulfide bridge patterns in vitro and in vivo. J Mol Biol. 2003;325:495–513.
Schmidt AM, Bloss I, Skerra A. Improved folding of apo-retinol-binding protein in the periplasm of Escherichia coli: positive influences of dsbC coexpression and of an amino acid exchange in the vitamin A binding site. Protein Eng. 1998;11:601–7.
Worley JR, Thompkins PB, Lee MH, Hutton M, Soloway P, Edwards DR, Murphy G, Knäuper V. Sequence motifs of tissue inhibitor of metalloproteinases 2 (TIMP-2) determining progelatinase A (proMMP-2) binding and activation by membrane-type metalloproteinase 1 (MT1-MMP). Biochem J. 2003;372:799–809.
Devy L, Dransfield DT. New strategies for the next generation of matrix-metalloproteinase inhibitors: selectively targeting membrane-anchored MMPs with therapeutic antibodies. Biochem Res Int. 2011;2011:191670.
Nam DH, Rodriguez C, Remacle AG, Strongin AY, Ge X. Active-site MMP-selective antibody inhibitors discovered from convex paratope synthetic libraries. Proc Natl Acad Sci USA. 2016;113:14970–5.
Imren S, Kohn DB, Shimada H, Blavier L, DeClerck YA. Overexpression of tissue inhibitor of metalloproteinases-2 retroviral-mediated gene transfer in vivo inhibits tumor growth and invasion. Cancer Res. 1996;56:2891–5.
Watanabe M, Takahashi Y, Ohta T, Mai M, Sasaki T, Seiki M. Inhibition of metastasis in human gastric cancer cells transfected with tissue inhibitor of metalloproteinase 1 gene in nude mice. Cancer. 1996;77:1676–80.
Khokha R, Zimmer MJ, Wilson SM, Chambers AF. Up-regulation of TIMP-1 expression in B16-F10 melanoma cells suppresses their metastatic ability in chick embryo. Clin Exp Metastasis. 1992;10:365–70.
Stricklin GP. Human fibroblast tissue inhibitor of metalloproteinases: glycosylation and function. Coll Relat Res. 1986;6:219–28.
Caterina NC, Windsor LJ, Bodden MK, Yermovsky AE, Taylor KB, Birkedal-Hansen H, Engler JA. Glycosylation and NH2-terminal domain mutants of the tissue inhibitor of metalloproteinases-1 (TIMP-1). Biochim Biophys Acta. 1998;1388:21–34.
Carmichael DF, Sommer A, Thompson RC, Anderson DC, Smith CG, Welgus HG, Stricklin GP. Primary structure and cDNA cloning of human fibroblast collagenase inhibitor. Proc Natl Acad Sci USA. 1986;83:2407–11.
Jackson HW, Defamie V, Waterhouse P, Khokha R. TIMPs: versatile extracellular regulators in cancer. Nat Rev Cancer. 2017;17:38–53.
Kim YS, Ahn YH, Song KJ, Kang JG, Lee JH, Jeon SK, Kim HC, Yoo JS, Ko JH. Overexpression and beta-1,6-N-acetylglucosaminylation-initiated aberrant glycosylation of TIMP-1: a “double whammy” strategy in colon cancer progression. J Biol Chem. 2012;287:32467–78.
Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, Panico M, Morris HR, Dell A, Wren BW, Aebi M. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science. 2002;298:1790–3.
Valderrama-Rincon JD, Fisher AC, Merritt JH, Fan YY, Reading CA, Chhiba K, Heiss C, Azadi P, Aebi M, DeLisa MP. An engineered eukaryotic protein glycosylation pathway in Escherichia coli. Nat Chem Biol. 2012;8:434–6.
Hayhurst A, Happe S, Mabry R, Koch Z, Iverson BL, Georgiou G. Isolation and expression of recombinant antibody fragments to the biological warfare pathogen Brucella melitensis. J Immunol Methods. 2003;276:185–96.
Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–30.
Segatori L, Paukstelis PJ, Gilbert HF, Georgiou G. Engineered DsbC chimeras catalyze both protein oxidation and disulfide-bond isomerization in Escherichia coli: reconciling two competing pathways. Proc Natl Acad Sci USA. 2004;101:10018–23.
Wang J, Petefish JW, Hillier AC, Schneider IC. Epitaxially grown collagen fibrils reveal diversity in contact guidance behavior among cancer cells. Langmuir. 2015;31:307–14.
Authors’ contributions
KBL, DHN, ICS, and XG designed the research; KBL, DHN, JAMN, and JW conducted the experiments; KBL, DHN, JAMN, JW, ICS and XG analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The figures and tables supporting the conclusions of this study are included in this published article and its supplementary information files.
Funding
This work was supported by National Science Foundation the Faculty Early Career Development (CAREER) Program 1453645 and National Institutes of Health Grant R01 GM 115672. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
12934_2017_686_MOESM1_ESM.docx
Additional file 1. Figure S1. Size exclusion chromatography of purified N-TIMP-1 prepared without DsbC co-expression. The size-exclusion column was equilibrated with 50 mM HEPES (pH 7.5) and 150 mM NaCl. 100 µL of 500 µg/mL N-TIMP-1 was loaded to a superdex™ 75 10/300 GL column (10 mm × 300 mm) at a flow rate of 0.5 mL/min. Chromatograms were obtained by monitoring absorbance at 280 nm. The molecular mass of N-TIMP-1 was estimated by its retention time and comparison with these of standard molecular mass markers, e.g. ovalbumin (43 kDa) and lysozyme (14.3 kDa).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lee, K.B., Nam, D.H., Nuhn, J.A.M. et al. Direct expression of active human tissue inhibitors of metalloproteinases by periplasmic secretion in Escherichia coli . Microb Cell Fact 16, 73 (2017). https://doi.org/10.1186/s12934-017-0686-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-017-0686-9