
Abstract
Background
Cyanobacterium Synechococcus elongatus PCC 7942 holds promise for biochemical conversion, but gene deletion in PCC 7942 is time-consuming and may be lethal to cells. CRISPR interference (CRISPRi) is an emerging technology that exploits the catalytically inactive Cas9 (dCas9) and single guide RNA (sgRNA) to repress sequence-specific genes without the need of gene knockout, and is repurposed to rewire metabolic networks in various procaryotic cells.
Results
To employ CRISPRi for the manipulation of gene network in PCC 7942, we integrated the cassettes expressing enhanced yellow fluorescent protein (EYFP), dCas9 and sgRNA targeting different regions on eyfp into the PCC 7942 chromosome. Co-expression of dCas9 and sgRNA conferred effective and stable suppression of EYFP production at efficiencies exceeding 99%, without impairing cell growth. We next integrated the dCas9 and sgRNA targeting endogenous genes essential for glycogen accumulation (glgc) and succinate conversion to fumarate (sdhA and sdhB). Transcription levels of glgc, sdhA and sdhB were effectively suppressed with efficiencies depending on the sgRNA binding site. Targeted suppression of glgc reduced the expression to 6.2%, attenuated the glycogen accumulation to 4.8% and significantly enhanced the succinate titer. Targeting sdhA or sdhB also effectively downregulated the gene expression and enhanced the succinate titer ≈12.5-fold to ≈0.58–0.63 mg/L.
Conclusions
These data demonstrated that CRISPRi-mediated gene suppression allowed for re-directing the cellular carbon flow, thus paving a new avenue to rationally fine-tune the metabolic pathways in PCC 7942 for the production of biotechnological products.
Similar content being viewed by others

Background
Cyanobacteria are photoautotrophic procaryotes that can exploit sunlight and CO2 as the sole energy and carbon sources to convert CO2 into organic compounds via photosynthesis [1]. Many cyanobacterial strains are amenable to natural transformation and homologous recombination for gene manipulation. Thanks to these attributes, genetically engineered cyanobacteria have drawn increasing attention as a chassis for the production of biofuels and bio-derived chemicals [2]. For instance, Synechococcus elongatus PCC 7942 has been genetically modified as a “cell factory” to divert native metabolic pathways to produce 2,3-butanediol, 2-methyl-1-butanol, isopropanol, free fatty acid, 1,2-propanediol, isopropanol, isobutyraldehyde and isobutanol, etc. [3].
Typically, PCC 7942 is engineered by knocking in genes encoding synthetic pathways and/or knocking out genes in competing pathways [4, 5]. However, generation of a single gene knockout mutant may take >3 weeks using conventional methods [6] due to its long doubling time and oligoploidy nature [1]. Sometimes deletion of certain genes essential for metabolic balances is not feasible or easily achieved as the deletion might be lethal to the cells. Furthermore, in many cases intermediate levels of enzyme expression may result in better product titer [7]. Therefore, tunable and balanced gene expression is desirable for high productivity, product titer, and conversion yield, and controllable gene repression/knockdown may be preferable than gene deletion for certain biotechnological applications and synthetic biological manipulations [8].
CRISPR-Cas9 is a newly developed RNA-guided genome editing system [9, 10]. CRISPR-Cas9 system comprises the Cas9 nuclease, transacting RNA (tracrRNA) and CRISPR RNA (crRNA). crRNA/tracrRNA complexes with Cas9 and, guided by the spacer sequence on crRNA, orchestrate to recognize protospacer-adjacent motif (PAM) and bind to proximal complementary sequence. After the recognition and binding, Cas9 nuclease triggers double strand break (DSB) at the chromosomal DNA [11]. Coupled with an editing template DNA, such CRISPR-Cas9-mediated DSB is exploited for programmable genome engineering of diverse cell types and model organisms [12–16], as well as for gene and cell therapy [17–19]. We have also employed CRISPR to engineer the PCC 7942 genome [20] and effectively inserted DNA fragments as large as 7 kb into Escherichia coli genome [21].
Furthermore, the catalytic domains of Cas9 are mutated to generate the inactive Cas9 (dCas9) lacking the endonuclease activity. dCas9 is used in conjunction with the chimeric single guide RNA (sgRNA) wherein the mature crRNA is fused to a partial tracrRNA to mimic the natural crRNA:tracrRNA duplex. By co-expression, the dCas9-sgRNA complex specifically binds to the target gene at the promoter or coding sequence and acts as a roadblock to the elongating RNA polymerase, hence aborting transcription initiation or elongation [22]. This new technology, termed CRISPR interference (CRISPRi), was recently repurposed to repress sequence-specific genes in diverse eucaryotic and procaryotic cells, for rewiring metabolic networks [23, 24] and high-throughput interrogation of genome-wide gene functions [25, 26]. Very recently, CRISPRi has also exploited for gene regulation in cyanobacteria Synechcocystis sp. PCC 6803 [27] and Synechococcus sp. PCC 7002 [7]. However, whether CRISPRi functions in PCC 7942 has yet to be explored.
In this study, we harnessed the CRISPRi system to effectively knockdown exogenous and endogenous genes in PCC 7942 via appropriate sgRNA design. Selective repression of glgc, sdhA and sdhB genes increased the succinate production by PCC 7942, hence demonstrating the feasibility of employing CRISPRi for the metabolic engineering of PCC 7942 and production of bio-derived chemicals.
Methods
Microorganisms
All molecular cloning experiments were performed using E. coli DH5α strain (Sigma). Unless otherwise noted, for suspension culture S. elongatus PCC 7942 (Invitrogen) was cultivated in a 250 ml shake flask containing 40 ml BG-11 medium [4, 5] with or without antibiotics (gyratory shaking at 100 rpm, with sterile air containing 0.04% CO2) in a 30 °C incubator (600SR, Hipoint) with illumination from continuous cool white fluorescent light (intensity ≈ 70 μmol//m2 s). For solid culture, PCC 7942 cells were streaked onto 90 mm plates containing 40 cm3 BG-11/agar medium supplemented with 1 mM sodium thiosulfate and appropriate antibiotic, and incubated with continuous illumination (intensity 70 µmol//m2 s) for 7–9 days until colonies developed.
Plasmids construction
pdCas9-bacteria plasmid (Addgene #44249) harbored chloramphenicol resistance gene (CmR) and dCas9 gene (derived from S. pyogenes) driven by the PLtetO1 promoter [24]. The sequences homologous to the 5′ (5-NSI) and 3′ (3-NSI) end of PCC 7942 NSI site (neutral site I), together with the intervening origin of replication (ori), were PCR-amplified from pSYN_1 plasmid (Invitrogen) with flanking AvrII and SpeI sites. pdCas9-bacteria and the PCR product were separately digested with AvrII/SpeI and ligated together (Additional file 1: Figure S1). The resultant pLtetO1-dCas9 contained the CmR and PLtetO1-dCas9 expression cassettes flanked by the 5-NSI and 3-NSI homology arms (Additional file 1: Figure S1).
We next PCR-amplified the Psmt promoter (including the promoter smtA and the repressor smtB) from the PCC 7942 chromosome, which was cloned into pLtetO1-dCas9 by AflII/BglII digestion to replace the PLtetO1 promoter, yielding pSdCas9 (Additional file 1: Figure S2). The eyfp gene under the control of PconII promoter was PCR-amplified from pconII-EYFP’ (see Additional file 1: Supplementary Methods) and subcloned into pSdCas9 by AvrII/SmaI (Additional file 1: Figure S3). The resultant pSdCas9-CY’ harbored the expression cassettes comprising CmR, dCas9 under Psmt and eyfp under PconII, which were flanked by homology arms targeting the NSI site (5-NSI and 3-NSI).
pgRNA-bacteria plasmid (Addgene, #44251) contained the ampicillin-resistance gene (ApR) and an sgRNA backbone driven by PJ23119 promoter. The sgRNA backbone comprised the base-pairing (spacer) region (20 bp), dCas9 handle (42 bp) and the S. pyogenes terminator (40 bp) as described [22]. To replace the spacer sequence on the sgRNA backbone with new spacer sequences targeting different regions on the PCC 7942 chromosome, we designed a reverse primer Ec_R and forward primers Ec_F with different new spacer sequences (Table 1), and performed inverse PCR (iPCR) using pgRNA-bacteria as the template [22]. The resultant PCR products comprising the new sgRNA sequences, ApR and PJ23119 promoter were phosphorylated using T4 polynucleotide kinase and joined using T4 DNA ligase to form new plasmids (bacteria sgRNA plasmids) containing sgRNA targeting different regions on the PCC 7942 chromosome (Additional file 1: Figure S4A).
To integrate the sgRNA sequences into NSII (neutral site II) site, the sequences homologous to the 5′ (5-NSII) and 3′ (3-NSII) end of PCC 7942 NSII site were PCR-amplified from pNSII_plus plasmid (kindly provided by Prof. James Liao). The resultant PCR product consisted of 5-NSII, origin of replication (ColE1), 3-NSII and kanamycin resistance gene (KmR). The PCR product and bacteria sgRNA plasmid were digested by EcoRI/BamHI and ligated to form the psgRNA plasmids as shown in Additional file 1: Figure S4B. The resultant plasmids were designated as psgRNA::Φ, psgRNA::P1, psgRNA::NT1, psgRNA::NT2, psgRNA::glgc1, psgRNA::glgc2, psgRNA::sdhA1, psgRNA::sdhA2, psgRNA::sdhB1 or psgRNA::sdhB2, depending on the target gene and location (see “Results” section).
Transformation and recombinant cell construction
For transformation into PCC 7942, 40 ml cells in the shake flask were cultured to optical density at 730 nm (OD730) = 0.6–0.8, centrifuged (5000×g for 15 min), washed with 20 ml BG-11 medium, centrifuged again, resuspended in 2 ml BG-11 medium and aliquoted (300 µl per microfuge tube). The plasmids were quantified using Nanodrop 2000 (Thermo), and 2000 ng plasmid was added to the tubes and mixed well with the aliquoted cells. The tubes were wrapped with foil and incubated with the rotary mixer in the incubator (30 °C) for 24 h to enhance the transformation efficiency as described [28]. The transformed cells were streaked onto the BG-11/agar plate containing appropriate antibiotics (e.g. 5 µg/ml Cm and 10 µg/ml Km) and cultured until colonies developed. The colonies were re-streaked twice onto the BG-11/agar plate containing antibiotics to yield the recombinant cells.
Growth curve, flow cytometry and confocal microscopy
The recombinant cells were transferred to and cultured in the shake flask containing Cm/Km. In parallel, wild-type (WT) cells were cultured in the same fashion without antibiotics. One milliliter of cells were sampled daily for the measurement of OD730 for the growth curve. The cells (1 ml) were also withdrawn every 3 days for EYFP analysis, followed by replenishment with 4 ml fresh BG-11 medium with or without antibiotics.
For eyfp expression analysis, the cells were subjected to flow cytometry (FACSCalibur, BD Biosciences) and the mean fluorescence intensity (FI) of 10,000 cells was measured. The mean FI (in arbitrary unit, a.u.) of each group was subtracted from that of WT cells to yield the final mean FI.
Alternatively, the eyfp-expressing recombinant cells were cultured in the shake flask to mid-log phase (OD730 = 1–1.5), and the cells were subjected to flow cytometry analysis or observed at 1000× under a confocal microscope (Eclipse TE2000-E, Nikon) for the yellow fluorescence (488 nm) and auto fluorescence (543 nm).
Quantification of mRNA by qRT-PCR
PCC 7942 cells were cultured in 40 ml BG-11 medium with or without antibiotics to OD730 = 0.7–1.3 and 5 ml cells were centrifuged (17,000×g for 5 min) and stored at −80 °C. After thawing, total RNA was extracted using NucleoSpin® RNAIIKit (Macherey Acherey-Nagel) with minor modifications to increase the extraction efficiency: the lysozyme concentration was increased to 2 mg/ml and reaction time at 37 °C was extended to 20 min. The extracted mRNA was quantified using a spectrophotometer (Nanodrop 2000, Thermo) and 1 µg RNA was reverse-transcribed to cDNA using the MMLV Reverse Transcription 1st-strand cDNA Synthesis Kit (Epicentre Biotechnologies). The cDNA was diluted in 1 ml deionized water and stored at −20 °C. After thawing, 3 µl cDNA was mixed with 1.5 µl deionized water, 0.5 µl gene-specific primer pairs (10 µM, Table S1) and 5 µl SYBR® Green PCR Master Mix (Applied Biosystems). Subsequent quantitative real-time PCR (qPCR) was performed using StepOnePlus™ (Applied Biosystems) with the PCC 7942 housekeeping gene rnpB as the internal control [29]. Gene expression levels in all groups were normalized to those in the WT control group.
Glycogen analysis
Because PCC 7942 accumulates glycogen under nitrogen starvation conditions [30], we cultured cells in nitrate-deplete (0×N) BG-11 medium (which is similar to BG-11 medium except that NaNO3 was not included) to force cells to accumulate glycogen. Recombinant PCC 7942 cells cultured in shake flasks to mid-log phase (OD730 ≈ 0.7–1.3) were centrifuged (25 °C, 6000×g for 20 min), resuspended in 20 ml nitrate-deplete (0×N) BG-11 medium, centrifuged again, resuspended in 40 ml nitrate-deplete BG-11 medium containing Km/Cm, and cultured for 2 more days. As a control, WT cells were cultured and washed in a similar fashion, and cultured in nitrate-deplete, antibiotic-free BG-11 medium for 2 more days.
The cell density was adjusted to OD730 = 1 and 1 ml of cells were centrifuged (17,000×g for 5 min), followed by resuspension in 1 ml deionized water and homogenization using Bead Beater (Kelowna). After centrifugation (17,000×g, 5 min), the glycogen content (µg) in the supernatant was analyzed using the Glycogen Colorimetric/Fluorometric Assay Kit (Biovision, k646-100).
Succinate analysis
Recombinant PCC 7942 cells cultured in the shake flasks to stationary phase (OD730 ≈ 2.0) were centrifuged (25 °C, 6000×g for 20 min), and resuspended in 20 ml nitrate-deplete (0×N) BG-11 medium. After centrifugation, the cells were resuspended in 40 ml nitrate-deplete BG-11 medium containing Km/Cm and cultured in the shake flasks.
As a control, WT PCC 7942 cells were cultured in 80 ml BG-11 medium to OD730 ≈ 2.0, divided in half into 50 ml tubes, washed twice by centrifugation (6000×g, 20 min) and resuspension in 40 ml complete or nitrate-deplete BG-11 medium, followed by shake flask culture using 40 ml complete (1×N) or nitrate-deplete (0×N) BG-11 medium.
After 2 days, 1.5 ml recombinant or WT cells were collected, filtered through 0.22 µm and the supernatant was analyzed by UFLC-MS (LCMS-2020, Shimadzu) with a column [Acclaim™ Organic Acid, 3 µm, 2.1 × 150 mm (Dionex)] for the succinic acid titer (mg/l).
Statistical analysis
All quantitative data were analyzed using student’s t test. All data represent the averages of at least 3 independent culture experiments. p < 0.05 was considered significant.
Results
Pre-test of promoters for gene expression in S. elongatus PCC 7942
Although a number of inducible promoters (e.g. high-light-responsive psbA promoter, copper-regulated petE promoter, nitrate/nitrite-inducible PnirA promoter and the nickel-regulated nrsA promoter) have been assessed in cyanobacteria, induction of these regulatory systems could concurrently affect the endogenous cognate regulon and result in unwanted effects [1, 31, 32]. To evaluate appropriate promoters useful for driving CRISPRi in PCC 7942, we compared different inducible promoters (Psmt, PLtetO1, PConII-ribo, Ptrc, PLlacO1 and PBAD) derived from E. coli or cyanobacteria (Additional file 1: Figure S5). We determined that Psmt gave the highest enhanced yellow fluorescent protein (EYFP) expression and the highest induction ratio in PCC 7942 (Additional file 1: Figure S6). We next assessed various constitutive promoters (Ptrc’, PLlacO1′, PconII, PJ23101 and PJ23119, Additional file 1: Figure S7) and determined that PconII and PJ23119 gave rise to the highest eyfp expression levels (Additional file 1: Figure S8).
Establishment of CRISPRi system in PCC 7942
In light of aforementioned data, these 3 promoters were chosen for subsequent establishment of CRISPRi system in PCC 7942. We constructed pSdCas9-CY’ with the expression cassette comprised of chloramphenicol resistance gene (CmR), dCas9 under Psmt, and eyfp under PconII, which was flanked by homology arms targeting the NSI site (Fig. 1a). In parallel, we constructed a series of psgRNA plasmids harboring the cassette expressing kanamycin resistance gene (KmR) and sgRNA under the PJ23119 promoter, which was flanked by NSII-targeting homology arms (upper panel, Fig. 1b). The sgRNA were designed to target the eyfp cassette at the non-template strand of promoter (P1) or coding regions near the transcription start site (NT1) or near the middle of gene (NT2) so that the fluorescence intensity served as the indicator of gene repression (lower panel, Fig. 1b). We transformed pSdCas9-CY’ into PCC 7942 for cassette integration into NSI site, re-streaked, and then transformed individual psgRNA into the recombinant cells for integration into NSII site (Additional file 1: Figure S9), yielding recombinant clones expressing EYFP, dCas9 and different sgRNA (dCas9::P1, dCas9::NT1 or dCas9::NT2). As controls, we also constructed cells that expressed (1) only EYFP and the sgRNA targeting P1 (P1 group); (2) dCas9 and EYFP (dCas9 group) and (3) dCas9, EYFP and a scramble sgRNA (dCas9::Φ group). The cells were streaked onto BG-11/agar plates containing Km/Cm for 7–9 days, and resistant (KmR/CmR) colonies were picked for colony PCR to verify correct integration (data not shown). The colonies were transferred to shake flasks and cultured to OD730 = 1–1.5 in BG-11 medium containing appropriate antibiotics.

Establishment of CRISPRi in PCC 7942. a Map of pSdCas9-CY’ harboring CmR, dCas9 under Psmt and eyfp under PconII, which was flanked by homology arms targeting the NSI site. Trrnb, transcriptional terminator. Psmt consists of the smtA promoter and the smtB repressor. b Map of psgRNA harboring the cassette expressing KmR and sgRNA under the PJ23119 promoter, which was flanked by NSII-targeting homology arms. The sgRNAs were designed to target no sequence on the PCC 7942 chromosome (Φ) or the eyfp cassette at the non-template strand of promoter (P1) or coding regions near the transcription start site (NT1 and NT2). The numbers indicate the position relative to the transcription start site. c Confocal micrographs of cells. d Flow cytometry analysis data. pSdCas9-CY’ was first transformed into PCC 7942 for cassette integration into NSI site, re-streaked, then individual psgRNA was transformed into the recombinant cells for integration into NSII site. The transformants colonies were transferred to shake flasks and cultured to OD730 = 1–1.5 in BG-11 medium containing appropriate antibiotics
The fluorescence micrographs (Fig. 1c) showed that all groups had similar auto-fluorescence and the 3 control groups (P1, dCas9 and dCas9::Φ) lacking complete functional dCas9/sgRNA complex exhibited similar EYFP expression. In contrast, EYFP expression was diminished in the 3 experimental groups (dCas9::P1, dCas9::NT1 and dCas9::NT2) expressing both dCas9 and sgRNA (Fig. 1c). The flow cytometry analysis (Fig. 1d) further depicted that EYFP expression in the dCas9::P1, dCas9::NT1 and dCas9::NT2 was suppressed to ≈4.5, ≈0.9 and ≈24.0% that of the control dCas9::Φ group, respectively. These data confirmed the successful establishment of CRISPRi system that effectively repressed gene expression for up to ≈111-fold in PCC 7942. It is noteworthy that dCas9 was driven by an inducible promoter Psmt which could be induced with ZnSO4. However, even without inducer, the EYFP repression was still very effective (Fig. 1c), suggesting that a low basal level of dCas9 expression was sufficient to mediate the gene suppression. Therefore, the inducer was not added in all subsequent experiments.
Effect of CRISPRi on PCC 7942 growth and persistent transgene repression
To examine whether CRISPRi conferred long-term, stable gene suppression and imposed toxicity, the KmR/CmR colonies of all groups were transferred to shake flasks and cultured in 40 ml BG-11 medium containing Km/Cm. As a control, wild-type (WT) cells were cultured in 40 ml BG-11 without antibiotics. The cells were cultured for 21 days, during which cells were sampled for OD730 or flow cytometry analysis. As judged from the flow cytometry analysis (Fig. 2a), dCas9::Φ continued to express EYFP throughout the experiment, whereas the EYFP expression in the dCas9::P1, dCas9::NT1 and dCas9::NT2 groups remained suppressed for 21 days, proving that CRISPRi was able to persistently knockdown gene expression in PCC 7942. Meanwhile, all groups, including the WT control, had virtually overlapped growth curves (p > 0.05, Fig. 2b), indicating that dCas9 expression and persistent sgRNA expression did not affect the cell growth.

Effect of CRISPRi on PCC 7942 growth and persistent transgene repression. a Mean fluorescence intensity (FI) profiles. b Growth curves. The recombinant cells were cultured in 40 ml BG-11 medium containing Km/Cm. Wild-type (WT) cells were cultured in 40 ml BG-11 without antibiotics. The cells were cultured for 21 days, during which cells were sampled for OD730 or flow cytometry analysis
CRISPRi-mediated suppression of endogenous genes
Cyanobacteria can accumulate glycogen as a carbon sink under nitrogen starvation conditions. Deleting the glgc gene can abolish glycogen synthesis [33, 34] because glgc gene product is a key enzyme in the glycogen synthesis pathway. Conversely, sdhA and sdhB gene products are responsible for converting succinate to fumarate in the TCA cycle.
To evaluate the ability of CRISPRi to suppress endogenous genes, we constructed pSdCas9 that expressed dCas9 under Psmt and a new series of psgRNA that targeted no sequences on the genome of PCC 7942 (psgRNA::Φ) or different coding regions of glgc, sdhA and sdhB genes (Fig. 3a). PCC 7942 cells were transformed with pSdCas9 first for integration into NSI site, and were transformed again with individual psgRNA plasmids for integration into NSII site. The KmR/CmR colonies were transferred to shake flasks and cultured to OD730 = 0.7–1.3, and 5 ml cells were sampled for mRNA analysis.

Schematic illustration of plasmids used for CRISPRi-mediated suppression of glgc, sdhA and sdhB. Φ indicates the scramble sgRNA. Glgc1, glgc2, sdhA1, sdhA2, sdhB1 and sdhB2 are the sgRNA targeting different regions of endogenous genes. pSdCas9 expressed dCas9 under Psmt
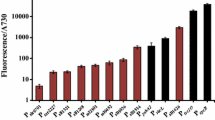
The qRT-PCR analysis (Fig. 4a–c), using the expression in WT cells as the basis, showed that the scramble sgRNA (Φ) did not significantly (p > 0.05) affect the expression of glgc, sdhA or sdhB. In contrast, the sgRNA targeting the non-template coding regions of glgc (glgc1 and glgc2) attenuated the glgc expression to ≈6.2 and ≈26.6%, respectively (Fig. 4a), which represented ≈16.1- and ≈3.8-fold repression, respectively. The sgRNA targeting sdhA (sdhA1 and sdhA2) and sdhB (sdhB1 and sdhB2) mitigated the expression to ≈18.9, ≈71.2, ≈33.1 and ≈36.6%, respectively (Fig. 4b, c). We also designed sgRNA targeting further downstream of the coding regions and found poorer repression efficiency (data not shown). These data attested that CRISPRi effectively suppressed the endogenous gene expression, and the silencing efficacy was inversely correlated with the distance of target region from the transcription start site.

Effective suppression of endogenous genes by CRISPRi. a glgc expression. b sdhA expression. c sdhB expression. PCC 7942 cells were transformed with pSdCas9 first for integration into NSI site, and were transformed again with individual psgRNA plasmids for integration into NSII site. The KmR/CmR colonies were transferred to 40 ml BG-11 medium containing Km/Cm and cultured to OD730 = 0.7–1.3, and 5 ml cells were sampled for qRT-PCR analysis. The expression levels were normalized to those in the WT cells
CRISPRi-mediated gene downregulation enhanced the production of chemicals
To evaluate the effect of repressing glgc, sdhA or sdhB genes, KmR/CmR cells were cultured to OD730 ≈ 0.7–1.3, centrifuged and cultured in shake flasks containing 40 ml nitrogen-deplete BG-11 (BG-11 devoid of NaNO3) for 2 days. Under nitrogen starvation conditions, suppression of glgc gene concomitantly mitigated the glycogen accumulation to ≈4.8% (glgc 1 group) and 25.5% (glgc2 group) that of the WT group (Fig. 5a), but did not induce chlorosis (a condition in which cells produce insufficient chlorophyll, data not shown). We also attempted to target other non-template coding regions of glgc further far away from the start codon than glgc1 and glgc2, which attenuated glycogen accumulation with varying degrees (from 37 to 72%, data not shown).

Effects of CRISPRi-mediated suppression on glycogen accumulation and succinate production. a Glycogen titer (µg/OD730). b Succinate titer (mg/l). For glycogen analysis, KmR/CmR cells were cultured to OD730 ≈ 0.7–1.3, centrifuged and cultured in shake flasks containing 40 ml nitrogen-deplete (0×N) BG-11 for 2 days and subjected to glycogen analysis. To analyze succinate titer, PCC 7942 cells cultured in shake flasks to stationary phase (OD73 ≈ 2.0) were centrifuged, and resuspended in 20 ml nitrate-free BG-11 medium. After centrifugation, the cells were resuspended in 40 ml nitrate-deplete (0×N) BG-11 medium containing Km/Cm and cultured in shake flasks. The WT cells were cultured in the nitrate-deplete (0×N) BG-11 or BG-11 (nitrate-replete) (1×N) medium
Moreover, we examined the succinate production by individually targeting the aforementioned gene regions and compared the succinate levels with those of the non-targeting control (Φ group) and WT groups under nitrate-replete (1×N) and nitrate-deplete (0×N) conditions (nitrogen-deplete BG-11 medium). Figure 5b delineates that WT cells cultured with nitrogen (1×N) did not produce appreciable amounts of succinate, yet produced slightly more succinate (≈0.05 mg/L) under nitrogen starvation (0×N) conditions. Repression with glgc1, sdhA1, sdhB1 and sdhB2 led to significantly elevated succinate titer when compared with WT cells and the cells without CRISPRi suppression (Φ group), reaching ≈0.26, ≈0.61, ≈0.58 and ≈0.63 mg/L, respectively (Fig. 5b). By suppressing sdhA or sdhB, the succinate titer (≈0.58–0.63 mg/L) was ≈12.5-fold higher than that in the WT cells. These data collectively confirmed that CRISPRi was able to suppress endogenous genes and enhanced succinate production in PCC 7942.
Discussion
CRISPRi holds great promise for a wide range of applications in microorganisms, including bacterial cell growth control [35], genetic screen [25, 36], synthetic biology module development [37, 38] or metabolic networks control in various microorganisms such as E. coli [24, 39, 40], mycobacteria [41], Bacillus subtilis [42], Corynebacterium glutamicum [43], Clostridium beijerinckii [44], yeast [45] and cyanobacteria [7]. In particular, a number of recent studies have exploited CRISPRi to regulate the metabolic pathways in E. coli for enhanced production of various biotechnological products including poly(3-hydroxybutyrate-co-4-hydroxybutyrate) [23], terpenoid [8], pinosylvin [46], flavonoid [47] and mevalonate [48]. Escherichia coli is a popular host as it has been extensively studied, grows fast, possesses a single chromosome and plenty of genetic engineering toolkits have been developed for E. coli engineering. Furthermore, E. coli allows the replication and continued presence of plasmids within the cells, which renders easy establishment and maintenance of CRISPRi elements (dCas9 and sgRNA) in E. coli.
In contrast, cyanobacteria grow relatively slowly and current knowledge and synthetic biology tool development for cyanobacteria lag far behind those for E. coli [49]. Furthermore, many cyanobacteria possess multiple copies of chromosomes and foreign genes need to be integrated and selected, hence making it much more labor-intensive and time-consuming to establish the CRISPRi system in cyanobacteria. To our best knowledge, only 2 very recent studies have employed CRISPRi to engineer cyanobacteria [7, 27]. Yao et al. explored CRISPRi in Synechcocystis sp. PCC 6803 to repress formation of carbon storage compounds polyhydroxybutryate and glycogen during nitrogen starvation [27]. Gordon et al. established the CRISPRi system in Synechococcus sp. PCC 7002 to repress synthesis of carboxysome essential for CO2 concentrating mechanism and downregulate a key node in nitrogen assimilation for enhanced lactate production [7].
In this study, we exploited the CRISPRi system to modulate the gene expression in another common model cyanobacterium PCC 7942, which remarkably differs from PCC 6803 and PCC 7002 in many aspects such as positions in the phylogenetic tree, genome size, chromosome copy number, doubling time and growth conditions [49]. To test the feasibility, we first screened a number of inducible and constitutive promoters that might function in PCC 7942. Based on our preliminary data (Additional file 1: Figures S5–S8), we selected the inducible promoter Psmt with lowest leaky expression and highest induction ratio to drive the dCas9 expression, as well as constitutive promoters PconII and PJ23119 to drive the eyfp and sgRNA expression, respectively (Fig. 1a, b). Notably, even without the addition of inducer, the dCas9/sgRNA still suppressed the EYFP expression (Fig. 1c, d). In accord with our observations, Yao et al. employed a panel of PL promoters to drive the dCas9 expression, which however, remarkably repressed the GFP expression in the absence of inducer [27]. Likewise, Gordon et al. employed an anhydrotetracycline (aTc)-inducible promoter to drive dCas9 expression and observed significant EYFP repression in the uninduced state [7]. These findings altogether indicate that leaky expression of dCas9 triggers robust gene perturbation and suggest that a low dCas9 level suffices to mediate CRISPRi-guided suppression.
Such leaky dCas9 expression gave rise to persistent EYFP knockdown (Fig. 2a) without appreciable negative effect on PCC 7942 growth (Fig. 2b), indicating that low level of dCas9 and continued expression of sgRNA do not compromise the health of PCC 7942. This attribute is desirable if the target gene is non-essential for cell growth/metabolism, because such persistent gene repression will obviate the need to add the inducer and save the cost in subsequent large-scale production process. However, if mitigating the target gene expression is detrimental to cell growth and function, an inducible promoter capable of stringent control is necessary. With this regard, Yao et al. have identified a tightly repressed promoter PL22 in PCC 6803 that allowed for efficient induction of dCas9 expression with minimal leaky dCas9 expression [27]. Conversely, Gordon et al. engineered the ribosome binding site (RBS) with reduced translation initiation efficiency so as to attenuate the leaky dCas9 expression in PCC 7002. Furthermore, a suite of homologous and heterologous inducible promoters (e.g. P idiA , P isiAB , PetE, PnrsB, etc.) have been developed/explored for their applicability in cyanobacteria [1, 31, 49, 50]. Future studies will be directed towards developing synthetic parts/circuits for tightly regulatable dCas9 expression and robust sgRNA expression in PCC 7942.
Meanwhile, here we designed sgRNA to target different regions of eyfp cassette and observed that targeting the regions near -35 of the promoter (P1) and the non-template coding sequence near the transcription start site (TSS) (NT1 and NT2) give rise to effective expression suppression. The data suggested that targeting the promoter and a region as close to the TSS as possible gave better suppression, which agreed well with the sgRNA design rule proposed previously for E. coli [22] and cyanobacteria PCC 6803 [27].
Given the successful repression of exogenous reporter gene, we further employed the same sgRNA design rule to selectively knockdown genes essential for glycogen accumulation (glgc) and succinate conversion to fumarate (sdhA and sdhB) (Fig. 3). Our data demonstrated successful downregulation of these genes using a single sgRNA targeting the coding regions, with sgRNA closer to the TSS resulting in more effective suppression (Fig. 4). In particular, repressing glgc using the glgc1 sgRNA effectively suppressed the glycogen accumulation (Fig. 5a) and ameliorated the succinate production titer (Fig. 5b). Conversely, glgc2 sgRNA, which targeted a region further downstream of the TSS gave rise to less effective glycogen accumulation and succinate production (Fig. 5). Likewise, repressing sdhA with different sgRNAs (sdhA1 and sdhA2) led to quite different degrees of sdhA downregulation (Fig. 4b) and sdhA1 conferred more effective sdhA suppression and higher succinate production (Figs. 4b, 5b). Conversely, suppressing sdhB with two different sgRNAs (sdhB1 and sdhB2) gave similar degrees of sdhB suppression and enhanced succinate production (Fig. 5b). These data altogether confirmed that CRISPRi is able to effectively and precisely suppress the target gene expression and re-direct carbon flux to the desired metabolic product, with positive correlation between the degrees of gene repression and succinate titer. Furthermore, the selected target gene and binding site could profoundly influence the outcome, hence underlining the importance of the sgRNA design.
Successful application of CRISPRi in PCC 7942 for gene knockdown is desirable, because PCC 7942 has been genetically engineered to divert native metabolic pathways for product formation [1, 2]. Traditional engineering approach involves the knockout of chromosomal genes, yet the oligoploidy nature of PCC 7942 makes it difficult to knockout the endogenous gene(s) on all chromosomes at once, hence necessitating time-consuming re-streaking and antibiotic selection. Moreover, knockout of certain genes essential for cell metabolism, survival and/or proper function will impair the ability of the cells to produce the desired product. CRISPRi offers the flexibility to fine-tune the endogenous gene expression levels without completely abrogating the gene functions, hence representing a valuable tool kit to intricately regulate the metabolic flux in the cells. Since CRISPRi can be used for genetic screen with appropriate sgRNA library design [25, 45], CRISPRi also may provide a promising tool to interrogate the functions of genes crucial for cell metabolism and product production in PCC 7942.
Conclusions
In summary, we demonstrated successful CRISPRi-mediated modulation of gene expression in the cyanobacterium S. elongatus PCC 7942. By appropriate sgRNA design, we were able to selectively knockdown exogenous reporter gene (eyfp) and endogenous genes (glgc, sdhA and sdhB). Targeted suppression of the endogenous genes involved in succinate synthesis increased the succinate production, with the product titer positively correlating with the degrees of gene suppression. These data demonstrated that CRISPRi enabled customizable RNA-guided, targeted gene suppression, which allowed for re-directing the cellular carbon flow. This study thus paves a new avenue to rationally fine-tune the metabolic pathways in PCC 7942 for the production of biotechnological products.
Abbreviations
- CRISPR:
-
clustered regularly interspaced short palindromic repeat
- CRISPRi:
-
CRISPR interference
- dCas9:
-
catalytically inactive Cas9
- sgRNA:
-
single guide RNA
- EYFP:
-
enhanced yellow fluorescent protein
- S. elongatus :
-
Synechococcus elongatus
- crRNA:
-
CRISPR RNA
- NSI:
-
neutral site I
- Km:
-
kanamycin
- Cm:
-
chloramphenicol
- Ap:
-
ampicillin
References
Zhou J, Zhu T, Cai Z, Li Y. From cyanochemicals to cyanofactories: a review and perspective. Microb Cell Fact. 2016;15:2.
Baroukh C, Munoz-Tamayo R, Steyer JP, Bernard O. A state of the art of metabolic networks of unicellular microalgae and cyanobacteria for biofuel production. Metab Eng. 2015;30:49–60.
Angermayr SA, Gorchs Rovira A, Hellingwerf KJ. Metabolic engineering of cyanobacteria for the synthesis of commodity products. Trends Biotechnol. 2015;33:352–61.
Shen CR, Liao JC. Photosynthetic production of 2-methyl-1-butanol from CO2 in cyanobacterium Synechococcus elongatus PCC7942 and characterization of the native acetohydroxyacid synthase. Energy Environ Sci. 2012;5:9574–83.
Atsumi S, Higashide W, Liao JC. Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat Biotechnol. 2009;27:1177–80.
Ramey CJ, Baron-Sola A, Aucoin HR, Boyle NR. Genome engineering in cyanobacteria: where we are and where we need to go. ACS Synth Biol. 2015;4:1186–96.
Gordon GC, Korosh TC, Cameron JC, Markley AL, Begemann MB, Pfleger BF. CRISPR interference as a titratable, trans-acting regulatory tool for metabolic engineering in the cyanobacterium Synechococcus sp. strain PCC 7002. Metab Eng. 2016;38:170–9.
Kim SK, Han GH, Seong W, Kim H, Kim S-W, Lee D-H, Lee S-G. CRISPR interference-guided balancing of a biosynthetic mevalonate pathway increases terpenoid production. Metab Eng. 2016;38:228–40.
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
He X, Tan C, Wang F, Wang Y, Zhou R, Cui D, You W, Zhao H, Ren J, Feng B. Knock-in of large reporter genes in human cells via CRISPR/Cas9-induced homology-dependent and independent DNA repair. Nucleic Acids Res. 2016;44:e85.
He ZY, Proudfoot C, Mileham AJ, McLaren DG, Whitelaw CBA, Lillico SG. Highly efficient targeted chromosome deletions using CRISPR/Cas9. Biotechnol Bioeng. 2015;112:1060–4.
Choi KR, Lee SY. CRISPR technologies for bacterial systems: current achievements and future directions. Biotechnol Adv. 2016;34:1180–209.
Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–78.
Ghosh S, Tibbit C, Liu JL. Effective knockdown of Drosophila long non-coding RNAs by CRISPR interference. Nucleic Acids Res. 2016;44:e84.
Xu L, Park KH, Zhao L, Xu J, El Refaey M, Gao Y, Zhu H, Ma J, Han R. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. 2016;24:564–9.
Maeder ML, Gersbach CA. Genome-editing technologies for gene and cell therapy. Mol Ther. 2016;24:430–46.
Muller M, Lee CM, Gasiunas G, Davis TH, Cradick TJ, Siksnys V, Bao G, Cathomen T, Mussolino C. Streptococcus thermophilus CRISPR-Cas9 systems enable specific editing of the human genome. Mol Ther. 2016;24:636–44.
Li H, Shen CR, Huang C-H, Sung L-Y, Wu M-Y, Hu Y-C. CRISPR-Cas9 for the genome engineering of cyanobacteria and succinate production. Metab Eng. 2016;28:293–302.
Chung M-E, Yeh I-H, Sung L-Y, Wu M-Y, Chao Y-P, Ng I-S, Hu Y-C. Enhanced integration of large DNA into E. coli chromosome by CRISPR/Cas9. Biotechnol Bioeng. 2016. doi:10.1002/bit.26056.
Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat Protocols. 2013;8:2180–96.
Lv L, Ren Y-L, Chen J-C, Wu Q, Chen G-Q. Application of CRISPRi for prokaryotic metabolic engineering involving multiple genes, a case study: controllable P(3HB-co-4HB) biosynthesis. Metab Eng. 2015;29:160–8.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–83.
Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CH, Koo BM, Marta E, et al. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell. 2016;165:1493–506.
Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17:5–15.
Yao L, Cengic I, Anfelt J, Hudson EP. Multiple gene repression in cyanobacteria using CRISPRi. ACS Synth Biol. 2016;5:207.
Lan EI, Chuang DS, Shen CR, Lee AM, Ro SY, Liao JC. Metabolic engineering of cyanobacteria for photosynthetic 3-hydroxypropionic acid production from CO2 using Synechococcus elongatus PCC 7942. Metab Eng. 2015;31:163–70.
Wang H, Yang H, Shivalila Chikdu S, Dawlaty Meelad M, Cheng Albert W, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8.
Hickman JW, Kotovic KM, Miller C, Warrener P, Kaiser B, Jurista T, Budde M, Cross F, Roberts JM, Carleton M. Glycogen synthesis is a required component of the nitrogen stress response in Synechococcus elongatus PCC 7942. Algal Res. 2013;2:98–106.
Ma AT, Schmidt CM, Golden JW. Regulation of gene expression in diverse cyanobacterial species by using theophylline-responsive riboswitches. Appl Environ Microbiol. 2014;80:6704–13.
Wang B, Wang J, Zhang W, Meldrum DR. Application of synthetic biology in cyanobacteria and algae. Front Microbiol. 2012;3:344.
Gründel M, Scheunemann R, Lockau W, Zilliges Y. Impaired glycogen synthesis causes metabolic overflow reactions and affects stress responses in the cyanobacterium Synechocystis sp. PCC 6803. Microbiology. 2012;158:3032–43.
Carrieri D, Paddock T, Maness P-C, Seibert M, Yu J. Photo-catalytic conversion of carbon dioxide to organic acids by a recombinant cyanobacterium incapable of glycogen storage. Energy Environ Sci. 2012;5:9457–61.
Wiktor J, Lesterlin C, Sherratt DJ, Dekker C. CRISPR-mediated control of the bacterial initiation of replication. Nucleic Acids Res. 2016;44:3801–10.
Singh AK, Carette X, Potluri LP, Sharp JD, Xu R, Prisic S, Husson RN. Investigating essential gene function in Mycobacterium tuberculosis using an efficient CRISPR interference system. Nucleic Acids Res. 2016;44:e143.
Didovyk A, Borek B, Hasty J, Tsimring L. Orthogonal modular gene repression in Escherichia coli using engineered CRISPR/Cas9. ACS Synth Biol. 2016;5:81–8.
Cress BF, Jones JA, Kim DC, Leitz QD, Englaender JA, Collins SM, Linhardt RJ, Koffas MA. Rapid generation of CRISPR/dCas9-regulated, orthogonally repressible hybrid T7-lac promoters for modular, tuneable control of metabolic pathway fluxes in Escherichia coli. Nucleic Acids Res. 2016;44:4472–85.
Lee YJ, Hoynes-O’Connor A, Leong MC, Moon TS. Programmable control of bacterial gene expression with the combined CRISPR and antisense RNA system. Nucleic Acids Res. 2016;44:2462–73.
Cui L, Bikard D. Consequences of Cas9 cleavage in the chromosome of Escherichia coli. Nucleic Acids Res. 2016;44:4243–51.
Choudhary E, Thakur P, Pareek M, Agarwal N. Gene silencing by CRISPR interference in mycobacteria. Nat Commun. 2015;6:6267.
Westbrook AW, Moo-Young M, Chou CP. Development of a CRISPR-Cas9 tool kit for comprehensive engineering of Bacillus subtilis. Appl Environ Microbiol. 2016;82:4876–95.
Cleto S, Jensen JVK, Wendisch VF, Lu TK. Corynebacterium glutamicum metabolic engineering with CRISPR interference (CRISPRi). ACS Synth Biol. 2016;5:375–85.
Wang Y, Zhang ZT, Seo SO, Lynn P, Lu T, Jin YS, Blaschek HP. Gene transcription repression in Clostridium beijerinckii using CRISPR-dCas9. Biotechnol Bioeng. 2016;113:2739–43.
Smith JD, Suresh S, Schlecht U, Wu M, Wagih O, Peltz G, Davis RW, Steinmetz LM, Parts L. St.Onge RP: Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design. Genome Biol. 2016;17:1–16.
J-l Liang, L-q Guo, J-f Lin, Z-q He, F-j Cai, J-f Chen. A novel process for obtaining pinosylvin using combinatorial bioengineering in Escherichia coli. World J Microbiol Biotechnol. 2016;32:1–10.
Wu J, Du G, Chen J, Zhou J. Enhancing flavonoid production by systematically tuning the central metabolic pathways based on a CRISPR interference system in Escherichia coli. Sci Rep. 2015;5:13477.
Li S, Jendresen CB, Grunberger A, Ronda C, Jensen SI, Noack S, Nielsen AT. Enhanced protein and biochemical production using CRISPRi-based growth switches. Metab Eng. 2016;38:274–84.
Berla BM, Saha R, Immethun CM, Maranas CD, Moon TS, Pakrasi HB. Synthetic biology of cyanobacteria: unique challenges and opportunities. Front Microbiol. 2013;4:246.
Markley AL, Begemann MB, Clarke RE, Gordon GC, Pfleger BF. Synthetic biology toolbox for controlling gene expression in the cyanobacterium Synechococcus sp. strain PCC 7002. ACS Synth Biol. 2015;4:595–603.
Authors’ contributions
CHH, CRS and YCH designed the project. CHH, HL, LYS and MYW performed the experiments. CHH and MYW analyzed the data. YCH wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors thank Prof. James Liao of University of California Los Angeles for the generous gift of pNSII_plus plasmid.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Additional methods and data are available in the Additional file 1.
Ethics approval and consent to participate
This manuscript dose not report data collected from humans or animals.
Funding
This work was supported by grants from the Ministry of Science and Technology, Taiwan (MOST 103B7028J2, 104-2622-8-007-001 and 105-2622-8-007-009).
Author information
Authors and Affiliations
Corresponding author
Additional file
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Huang, CH., Shen, C.R., Li, H. et al. CRISPR interference (CRISPRi) for gene regulation and succinate production in cyanobacterium S. elongatus PCC 7942. Microb Cell Fact 15, 196 (2016). https://doi.org/10.1186/s12934-016-0595-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-016-0595-3