
Abstract
Introduction
Cachexia contributes to increased mortality and reduced quality of life in Chronic Obstructive Pulmonary Disease (COPD) and may be associated with underlying gene expression changes. Our goal was to identify differential gene expression signatures associated with COPD cachexia in current and former smokers.
Methods
We analyzed whole-blood gene expression data from participants with COPD in a discovery cohort (COPDGene, N = 400) and assessed replication (ECLIPSE, N = 114). To approximate the consensus definition using available criteria, cachexia was defined as weight-loss > 5% in the past 12 months or low body mass index (BMI) (< 20 kg/m2) and 1/3 criteria: decreased muscle strength (six-minute walk distance < 350 m), anemia (hemoglobin < 12 g/dl), and low fat-free mass index (FFMI) (< 15 kg/m2 among women and < 17 kg/m2 among men) in COPDGene. In ECLIPSE, cachexia was defined as weight-loss > 5% in the past 12 months or low BMI and 3/5 criteria: decreased muscle strength, anorexia, abnormal biochemistry (anemia or high c-reactive protein (> 5 mg/l)), fatigue, and low FFMI. Differential gene expression was assessed between cachectic and non-cachectic subjects, adjusting for age, sex, white blood cell counts, and technical covariates. Gene set enrichment analysis was performed using MSigDB.
Results
The prevalence of COPD cachexia was 13.7% in COPDGene and 7.9% in ECLIPSE. Fourteen genes were differentially downregulated in cachectic versus non-cachectic COPD patients in COPDGene (FDR < 0.05) and ECLIPSE (FDR < 0.05).
Discussion
Several replicated genes regulating heme metabolism were downregulated among participants with COPD cachexia. Impaired heme biosynthesis may contribute to cachexia development through free-iron buildup and oxidative tissue damage.
Similar content being viewed by others


Background
Chronic Obstructive Pulmonary Disease (COPD) is a leading causes of death worldwide, and it maintains increasing morbidity and mortality [1]. COPD is diagnosed based on reduced, irreversible lung function however, comorbidities are common with COPD and negatively impact survival [2]. Muscle wasting and cachexia are relevant comorbidities among COPD patients with prevalence increasing with stage of illness [2]. Cachexia in COPD may be associated with underlying changes in gene expression that could provide valuable insights for surveillance and drug development.
Collectively, there have been limited investigations examining differential gene expression in the context of COPD cachexia. Previous studies of gene expression changes in COPD cachexia have had small sample sizes, non-disease controls for comparison, employed a candidate-gene approach, or have focused on proxies for cachexia such as low fat-free mass index (FFMI) [3, 4]. Nonetheless, these studies have highlighted interesting genes with relevant biology to cachexia such as FOXO1 and FOXO3 [5]. The candidate-gene expression approach is limited to our understanding of known biology. In contrast, the genome-wide method is an agnostic, hypothesis-free approach, that has elucidated novel molecular mechanisms of COPD pathogenesis [6]. A challenge for implementing the genome-wide approach is that a large number of study participants are typically required to achieve a genome-wide level of significance. Although low FFMI and low BMI are components of the multifactorial definition of cachexia, these measures alone are not sufficient to monitor cachexia in the entire population at risk [7].
Our goal was to identify and explore differentially expressed genes associated with COPD cachexia. To achieve our objective, we utilized genome-wide expression data to conduct differential gene expression analyses aimed at deciphering the molecular mechanisms associated with cachexia in COPD patients from COPDGene [8]. We then tested whether significantly differentially expressed genes would be replicated in a second cohort of COPD patients from the ECLIPSE study [9].
Methods
Study design and ethics
Institutional Review Board approval for all analyses was obtained from the University of Alabama at Birmingham and have been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All subjects included in this analysis provided written informed consent prior to their inclusion in the respective studies mentioned below. All statistical analyses were performed in R (version 3.5.3).
Study populations
For both cohorts, COPD was defined as airflow obstruction using post-bronchodilator spirometry as GOLD grade 2 (FEV1/FVC < 0.7 and 50% < FEV1 < 80% predicted; FEV1: forced expiratory volume in one second, FEV1/FVC: FEV1 expressed as a percentage of forced vital capacity) or higher [10]. Discovery analyses were performed in 400 participants with COPD from the COPDGene study (NCT00608764; www.copdgene.org). COPDGene is a multicenter, observational study intent on characterizing progression of and genetic susceptibility to COPD, the details of which have been described previously [8]. In brief, COPDGene enrolled Non-Hispanic White and African American current or former smokers, with at least a 10 pack-year smoking history, aged 45 to 80 years to follow longitudinally, with two visits 5 years apart. RNA samples were collected at the five-year follow-up visit. Replication analyses were performed in 114 participants with GOLD 2–4 COPD enrolled in the ECLIPSE (Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints; SCO104960, NCT00292552; www.eclipse-copd.com) cohort. In ECLIPSE, phenotype data was obtained via the database of Genotypes and Phenotypes (dbGaP accession: phs001252.v1.p1) and gene expression data from the Gene Expression Omnibus (GEO: GSE76705). In brief, ECLIPSE [9] was a three-year observational study of subjects with COPD aged 45 to 75 years with a smoking history of ten or more pack-years. Like COPDGene, smaller numbers of control subjects were enrolled in ECLIPSE, who were not included in analysis. Forty-six centers across twelve countries collected data at baseline, 3 months, 6 months, and then every 6 months for three years. The main goal of ECLIPSE was to identify biomarkers associated with the progression of disease severity.
Phenotype data
In COPDGene, cachexia was defined at visit two, approximately five years from baseline, as either self-reported, unintentional weight loss greater than 5% in the past year or low BMI (< 20 kg/m2) in addition to one of three criteria: low 6MWD (< 350 m; a surrogate for decreased muscle strength), anemia (hemoglobin< 12 g/dl) or low FFMI (FFMI < 15 kg/m2 among women and FFMI < 17 kg/m2 among men). Low 6MWD and anemia were classified using visit two data. FFMI was derived from an algorithm incorporating pectoralis muscle area on baseline chest computed tomography scans [11].
In ECLIPSE, cachexia was defined according to the Evans consensus definition [12] at year 1 of the study as: weight loss greater than 5 % in the past 12 months or low BMI (< 20 kg/m2) and at least three of the five criteria: low six-minute walking distance (a surrogate for decreased muscle strength), anorexia, abnormal biochemistry (anemia or high CRP), fatigue, and low FFMI [12]. FFM was measured using bioelectrical impedance. Low FFMI was defined as < 15 kg/m2 among women and < 17 kg/m2 among men. Abnormal biochemistry was defined as having anemia (hemoglobin< 12 g/dl) or high CRP (> 5 mg/l). Six-minute walking distance (6MWD) < 350 m [13] was used to classify decreased muscle strength [13]. Anorexia and fatigue were only available at baseline and end of study. Anorexia was classified if a participant reported not feeling like eating for 3 or more days in the week prior to either the baseline or 3-year follow-up visit. Fatigue was defined at either baseline or 3-year visits as a FACIT (Function Assessment of Chronic Illness Therapy) questionnaire score less than 34 [14].
Gene expression data
In COPDGene, RNA-sequencing was performed using whole-blood samples collected at visit 2 (~Year 5) from 400 current or former smokers with COPD. RNA-sequencing alignment and quality control was previously describe in detail [6]. Transcripts were included in analyses if expressed at levels > 1 count per million mapped reads in > 10 subjects. Potential batch effects and unknown artifacts were removed using the SVAseq R package [15] to estimate surrogate variables (SV’s) while accounting for the presence of known covariates: age, sex, library construction batch, and white blood cell (WBC) count percentages. Principle component analysis (PCA) was performed on SV residualized expression data to ensure minimal systematic bias (Supplementary Figure 1).
In ECLIPSE, normalized gene expression data was obtained from GEO (GEO: GSE76705). Sample preparation and quality control for gene expression data obtained from ECLIPSE have been previously described [16, 17]. Briefly, gene expression in ECLIPSE was obtained from whole-blood samples collected at baseline and profiled using the Affymetrix Human U133 Plus2 array. Gene expression data were log-transformed, normalized, then background corrected through multi-array averaging and quantile normalization using the affy Bioconductor package [18]. Probes with low variance (< 50%) and those which did not annotate within a specific gene were removed, leaving a dataset of 21,156 probes for analysis in 114 COPD cases. The amount of missing data for each probe was assessed for quality control, and no probes were removed due to missingness, defined as missing in more than 3% of samples.
Differential gene expression analysis
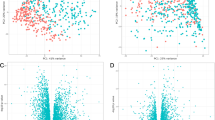
Differential gene expression analysis comparing cachectic and non-cachectic COPD patients from COPDGene was performed using the voom [19] and limma [20] R packages. Linear regression models were fit for each transcript and adjusted to control for age, gender, white blood cell (WBC) percentages, and significant SVs (N = 27). Significance for differentially expressed transcripts in COPDGene was defined as an FDR p-value < 0.05 adjusted for number of transcripts (Fig. 1).

Volcano plot of differential gene expression of cachectic vs. non-cachectic COPD patients from COPDGene. The plot shows the log2 fold change on the x-axis and the unadjusted p-value on the y-axis (on the -log10 scale). Blue dots represent genes with a fold changed greater than or equal to + 1 and a p-value < 0.05
In ECLIPSE, only probes mapping to a gene that was significantly differentially expressed in COPDGene were evaluated using linear regression models. A P < 0.05 was considered to be a significant replication in ECLIPSE. Models were adjusted to control for age, gender, and WBC percentages. In instances were multiple probes mapped to a single gene, the probe with the highest mean expression value was selected [21].
Gene overlap enrichment between genes significantly differentially expressed in COPDGene and ECLIPSE were calculated using the GeneOverlap R package [22] using a two-tailed Fisher’s exact to calculate the FDR corrected p-value between overlapping genes.
Pathway analysis
Gene set enrichment analysis (GSEA) of genes significantly differentially expressed in COPDGene and for those that replicated in ECLIPSE was performed using the Molecular Signatures Database (MSigDB) v6.1 [23]. This included gene set collections comprised of the hallmark gene set (N = 50 gene sets), curated gene sets (N = 4762 gene sets), Gene Ontology (GO) gene sets (N = 5917 gene sets), oncogenic signatures (N = 189 gene sets), and immunogenic signatures (N = 4872 gene sets). GSEA significance was defined as having an FDR-q p-value less than 0.05.
Results
Demographics
The prevalence of cachexia was 13.7% in COPDGene participants with COPD (Table 1).
There was no significant difference between the cachectic and non-cachectic COPD participants in COPDGene in terms of age (Table 1). On average, cachectic COPD subjects were more likely to be female (52.7% vs. 37.7%, p = 0.049), have lower lung function (FEV1% predicted 44.0 + 18.1 vs. 54.0 + 18.1, p < 0.001), and had less intensive smoking histories (46.8 + 19.2 vs. 53.5 + 26.0 pack years, p = 0.027) (Table 1). Cachectic COPD participants had lower BMI compared to non-cachectic COPD participants (22.0 + 4.9 vs 28.8 + 6.0, p < 0.001) (Table 1). As replication analyses were performed using data from the ECLIPSE study, we compared the characteristics of COPD participants included in the analyses in both studies in terms of age, sex, smoking duration, lung function, and BMI (Table 2). There were no significant differences between participants in COPDGene and ECLIPSE in terms of cumulative smoking exposure, lung function (FEV1 percent predicted), and BMI (Table 2). COPD participants in ECLIPSE were slightly younger (64.9 ± 5.3 vs. 68.7 ± 8.2 years, p < 0.05).
Differential gene expression in COPDGene
In COPDGene, 23 transcripts were significantly differentially expressed between cachectic and non-cachectic COPD participants (Table 2). Of these genes, 22 were downregulated among those with COPD cachexia.
Replication in ECLIPSE
In ECLIPSE, only 14 of the 23 significantly differentially expressed genes in COPDGene were measured on the Affymetrix expression array. All of which were also significantly differentially expressed among participants with COPD cachexia in ECLIPSE (Table 3). These genes include ALAS2, ANK1, ASCC2, CDC34, GUCD1, PLEK2, TNS1, SPTB, TRIM58, RILP, PPP2R5B, SMIM24, SLC25A39, and UBXN6 (Table 3). In addition, gene set overlap enrichment analysis of significantly differentially expressed genes in COPDGene and ECLIPSE identified these 14 genes as having a significant overlap (FDR-p = 7.9 × 10− 11).
Pathway analysis
The 23 differentially expressed genes associated with COPD cachexia in the discovery cohort were significantly enriched for gene sets involved in heme metabolism (Hallmark Heme Metabolism, FDR p-value = 5.92 × 10− 7) and heme biosynthesis (GO Heme Biosynthetic Process, FDR p-value = 3.85 × 10− 2), among others (Table S2). The 14 significantly differentially expressed COPD cachexia genes in the discovery which replicated were enriched with genes involved in heme metabolism (Hallmark Heme Metabolism, FDR-p = 4.50 × 10− 8; ALAS2, ANK1, TNS1, SPTB, TRIM58, PPP2R5) and heme biosynthesis (GO Heme Biosynthetic Process, FDR-p = 2.45 × 10− 2; ALAS2, SLC25A39), among others (Table 4). ASCC2, CDC34, UBXN6, PLEK2, RILP, SMIM24 and GUCD1 were not enriched in any significant pathways.
Discussion
In this study, we identified novel gene expression signatures associated with COPD cachexia in two independent cohorts of participants with moderate to severe COPD. All significant genes were downregulated among cachectic COPD patients. Several of these genes are involved in the regulation of heme metabolism (ALAS2, ANK1, TNS1, SPTB, TRIM58, PPP2R5) and biosynthesis (ALAS2 and SLC25A39). These findings may have implications for cachexia disease surveillance and drug development. Impaired heme metabolism and biosynthesis, decreased hepcidin production, and hemolysis may contribute to COPD cachexia through the induction of iron overloading leading to oxidative tissue damage, mitochondrial dysfunction, and aberrant repair (Fig. 2).

Summary of key findings in relationship to a putative role of iron in COPD cachexia. Genes in blue are significantly downregulated in COPD cachexia in manuscript analyses
Disruption of iron homeostasis in lung disease overall has received attention as a potential pathobiological mechanism in COPD [24,25,26]. Recently, Sato et al. [26] reported mice fed an iron deficient diet were more susceptible to cigarette smoke developing severe emphysema and lung hyperinflation. This finding conflicts with an earlier report by Cloonan et al. [25] where mice fed an iron deficient diet or given an iron chelator were less susceptible to cigarette smoke exposure. In keeping with Cloonan et al. [25], our findings may point to a role for iron toxicity through impaired heme biosynthesis and metabolism as a number of genes involved with these processes were downregulated in COPD cachexia (Fig. 2). Heme is an essential component of hemoglobin and anemia is a component of the criteria used to define cachexia. Anemia is a common co-morbidity in COPD [24]. We performed a sensitivity analysis to evaluate whether the 23 genes significantly differentially expressed among subjects with COPD cachexia were also associated with anemia and found no significant association (data not shown). A genome-wide expression study of skeletal muscle biopsies from upper gastro-intestinal cancer patients highlighted ALAS1 as downregulated in patients with cancer cachexia [27]. ALAS2 and ALAS1 encode the same enzyme, 5′-Aminolevuilnnate Synthetase, which plays a key role in heme synthesis [28]. Previous studies have demonstrated that ALAS1 and ALAS2 expression levels are increased during erythropoiesis in healthy individuals [28]. ALAS2 is erythroid-specific whereas ALAS1 is expressed in many tissues, including skeletal muscle. This may indicate defective heme biochemistry has ramifications outside of circulating red blood cells which could be relevant to both cancer and COPD cachexia. SLC25A39 encodes the solute carrier family 25 member 39, a mitochondrial membrane protein which plays a role in iron homeostasis and metabolism [29]. Specifically, SLC25A39 is involved in iron transportation and transfer, and interruptions in SLC25A39 biogenic activity have been shown to result in iron overloading [29]. ANK1 (encodes ankyrin 1) and SPTB (encodes the erythrocytic protein spectrin beta) are involved in erythrocyte membrane stability. Mutations in ANK1 and SPTB have been linked to erythrocyte membrane disorders including hereditary spherocytosis, in which clinically significant iron overload is commonly seen [30]. PPP2R5B (protein phosphatase 2 regulatory subunit B’beta) and TRIM58 (tripartite motif containing 58) function as regulators of erythropoiesis and mutations in these genes can lead to defective erythropoiesis and [31, 32] iron overloading [33]. TNS1 (Tensin 1) plays a role in heme metabolism and functions in the formation of fibrillar adhesion in muscle. Previous studies have identified an association between genetic variants in TNS1 and elevated risk of COPD and lung function decline [34]. Transcriptomic analysis has identified TNS1 as downregulated and differentially expressed in hypoxic alveolar cells [35]. Lung parenchymal destruction in COPD makes it difficult for the body to take in sufficient oxygen and can lead to hypoxemia (low blood oxygen saturation). Hypoxemic COPD patients often present with polycythemia, which suggests increased production of erythrocytes may be one mechanism for the body to maximize delivery of oxygen from the lungs [36]. Downregulation of TNS1 and other genes playing a critical role in heme metabolism could inhibit the body’s ability to effectively sequester the elevated levels of iron produced leading to an excess of iron.
In our study, PLEK2 was significantly downregulated in cachectic COPD patients. PLEK2 encodes pleckstrin-2, a protein playing a critical role in the protection of erythroblasts from oxidative damage [37]. Specifically, PLEK2 functions in the structural organization of the erythroblast to prevent the localization of cofilin (an actin-binding protein) to the mitochondria where it induces apoptosis [28, 37]. Previous studies have demonstrated pleckstrin-2 knockout results in erythrocyte apoptosis and inhibition of differentiation, a precursor to cell-free iron loading [37]. Our study indicates downregulation of PLEK2 could result in increased rates of hemolysis resulting in iron overloading (Fig. 2).
Hepcidin is essential for sequestering iron in the body and downregulation of hepcidin has been shown to cause severe iron overloading [38]. One gene, GUCD1, was downregulated in cachectic COPD patients. GUCD1 (guanylyl cyclase domain containing 1) is a ubiquitous protein that plays a role in regulating the growth of liver cells [39]. Hepcidin, produced in liver cells, degrades ferroprotein, which is responsible for releasing iron from cells [38]. Downregulation of GUCD1 in our study may suggest one mechanism of iron-overloading due to impaired hepcidin production (Fig. 2).
Iron overload due to inhibited iron metabolism has been implicated in increased production of reactive oxygen species, mitochondrial dysfunction, and oxidative stress [24]. Interestingly, mitochondrial dysfunction is one mechanism thought to contribute to the development of muscle wasting and dysfunction in COPD via buildup of oxidative stress [2]. In our study, the downregulation of genes involved in heme metabolism and biosynthesis, hepcidin production, and erythrocyte membrane stability may contribute to iron-overloading. Iron overloading has been implicated in the generation of mitochondrial dysfunction and oxidative damage, which are hallmarks of COPD cachexia pathogenesis (Fig. 2) [40].
Our study has several strengths and limitations. A strength of our study is that it presents the largest genome-wide expression study of cachexia in patients with COPD. We took advantage of a genome-wide approach which highlighted new candidate genes associated with COPD cachexia such as those involved with heme biochemistry. Another strength of our study was our ability to phenotype cachexia in our replication cohort (ECLIPSE) according to the criteria from the cachexia consensus definition [12], whereas previous studies have focused on COPD cachexia-associated proxy traits such as low FFMI or low BMI that lack specificity. Finally, we were also able to replicate 14 genes as a potential markers of COPD cachexia in an independent cohort of participants with COPD. Among the limitations, we were not able to test whether all 23 significantly differentially regulated genes in COPDGene replicated in ECLIPSE due to limitations with the microarray not having probes for all 23 genes. It is possible some of these genes would also replicate but requires further study in a COPD cohort with RNA-sequencing data. There was also limited phenotype data available to characterize cachexia in COPDGene to code the 5 criteria of the consensus definition; thus, we used the maximal set available. As not all criteria were available to categorize cachexia using the Evans consensus definition, participants with COPD may have been miscategorized. However, the criteria we used to classify cachexia in ECLIPSE were much more stringent and we replicated 14/23 findings. Further, unintentional weight loss in COPDGene was self-reported, which can be unreliable. We also dichotomized 6MWD (6MWD < 350 m) as a surrogate for muscle strength which could have misclassified some participants. Although it is reasonable to infer participants with very low 6MWD have low muscle strength, other factors such as cardiac and pulmonary function may affect 6MWD. We also analyzed gene expression data generated at a single time point. A longitudinal analysis would have provided a deeper and more powerful understanding of the changes in gene expression over time. Future analyses could prove useful in understanding the changes that occur over time in cachectic COPD patients and even prior to the development of cachexia to define at-risk populations. Also, we did not use qRT-PCR to validate significant findings from COPDGene However, our discovery analyses were performed with a fairly large sample size of 400 COPD subjects with replication in an independent cohort of COPD subjects. Finally, peripheral blood was used to evaluate gene expression in cachectic COPD subjects as opposed to skeletal muscle. However, our study replicates abovementioned findings using skeletal muscle biopsies to evaluate gene expression in patients with cancer cachexia [27]. Additionally, a peripheral blood sample is less invasive than a skeletal muscle biopsy, an important factor to consider due to the advanced age and accompanying frailty in COPD patients [1].
Conclusions
In conclusion, this study demonstrates the utility of genome-wide expression analyses to identify novel molecular genes and pathways associated with COPD cachexia. Specifically, our results indicated genes involved in heme biosynthesis and metabolism were significantly downregulated in cachectic patients with COPD. These results highlight impaired heme metabolism and biosynthesis as potential mechanisms for COPD cachexia pathogenesis via free iron build up, oxidative tissue damage, and aberrant repair. Further studies are warranted to determine potential efficacy of therapeutic targeting to prevent, attenuate, or even possibly reverse COPD cachexia.
Availability of data and materials
The datasets generated and/or analyzed during the current study for are available from dbGaP and GEO on reasonable request. [https://www.ncbi.nlm.nih.gov/geo/,
References
Marengoni A, Vetrano DL, Manes-Gravina E, Bernabei R, Onder G, Palmer K. The relationship between COPD and frailty: a systematic review and meta-analysis of observational studies. Chest. 2018;154(1):21–40.
Remels AH, Gosker HR, Langen RC, Schols AM. The mechanisms of cachexia underlying muscle dysfunction in COPD. J Appl Physiol (1985). 2013;114(9):1253–62.
Rabinovich RA, Drost E, Manning JR, Dunbar DR, Díaz-Ramos M, Lakhdar R, et al. Genome-wide mRNA expression profiling in vastus lateralis of COPD patients with low and normal fat free mass index and healthy controls. Respir Res. 2015;16:1.
Lewis A, Lee JY, Donaldson AV, Natanek SA, Vaidyanathan S, Man WD, et al. Increased expression of H19/miR-675 is associated with a low fat-free mass index in patients with COPD. J Cachexia Sarcopenia Muscle. 2016;7(3):330–44.
Doucet M, Russell AP, Léger B, Debigaré R, Joanisse DR, Caron MA, et al. Muscle atrophy and hypertrophy signaling in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176(3):261–9.
Parker MM, Chase RP, Lamb A, Reyes A, Saferali A, Yun JH, et al. RNA sequencing identifies novel non-coding RNA and exon-specific effects associated with cigarette smoking. BMC Med Genet. 2017;10(1):58.
McDonald MN, Wouters EFM, Rutten E, Casaburi R, Rennard SI, Lomas DA, et al. It's more than low BMI: prevalence of cachexia and associated mortality in COPD. Respir Res. 2019;20(1):100.
Regan EA, Hokanson JE, Murphy JR, Make B, Lynch DA, Beaty TH, et al. Genetic epidemiology of COPD (COPDGene) study design. COPD. 2010;7(1):32–43.
Vestbo J, Anderson W, Coxson HO, Crim C, Dawber F, Edwards L, et al. Evaluation of COPD longitudinally to identify predictive surrogate end-points (ECLIPSE). Eur Respir J. 2008;31(4):869–73.
Vestbo J, Hurd SS, Agustí AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–65.
McDonald MN, Diaz AA, Rutten E, Lutz SM, Harmouche R, San Jose Estepar R, et al. Chest computed tomography-derived low fat-free mass index and mortality in COPD. Eur Respir J. 2017;50(6)1701134. https://doi.org/10.1183/13993003.01134-2017.
Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr. 2008;27(6):793–9.
Cote CG, Casanova C, Marín JM, Lopez MV, Pinto-Plata V, de Oca MM, et al. Validation and comparison of reference equations for the 6-min walk distance test. Eur Respir J. 2008;31(3):571–8.
Van Belle S, Paridaens R, Evers G, Kerger J, Bron D, Foubert J, et al. Comparison of proposed diagnostic criteria with FACT-F and VAS for cancer-related fatigue: proposal for use as a screening tool. Support Care Cancer. 2005;13(4):246–54.
Leek JT. svaseq: removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 2014;42(21). https://doi.org/10.1093/nar/gku864.
Chang Y, Glass K, Liu YY, Silverman EK, Crapo JD, Tal-Singer R, et al. COPD subtypes identified by network-based clustering of blood gene expression. Genomics. 2016;107(2–3):51–8.
Singh D, Fox SM, Tal-Singer R, Bates S, Riley JH, Celli B. Altered gene expression in blood and sputum in COPD frequent exacerbators in the ECLIPSE cohort. PLoS One. 2014;9(9):e107381.
Gautier L, Cope L, Bolstad BM, Irizarry RA. Affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15.
Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29.
Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21(9):2067–75.
Miller JA, Cai C, Langfelder P, Geschwind DH, Kurian SM, Salomon DR, et al. Strategies for aggregating gene expression data: the collapseRows R function. BMC Bioinformatics. 2011;12:322.
Shen L. GeneOverlap: Test and visualize gene overlaps. R package version 1.22.0 ed; 2019.
Liberzon A. A description of the molecular signatures database (MSigDB) web site. Methods Mol Biol. 2014;1150:153–60.
Cloonan SM, Mumby S, Adcock IM, Choi AMK, Chung KF, Quinlan GJ. The "Iron"-y of Iron overload and Iron deficiency in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;196(9):1103–12..
Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabón MA, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22:163–74.
Sato K, et al. Effect of Iron Deficiency on a Murine Model of Smoke-Induced Emphysema. Am J Respir Cell Mol Biol. 2019. https://doi.org/10.1165/rcmb.2018-0239OC.
Stephens NA, Gallagher IJ, Rooyackers O, Skipworth RJ, Tan BH, Marstrand T, et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med. 2010;2(1):1.
Mello FV, Land MGP, Costa ES, Teodósio C, Sanchez ML, Bárcena P, et al. Maturation-associated gene expression profiles during normal human bone marrow erythropoiesis. Cell Death Discov. 2019;5:69.
Rouault TA, Tong WH. Tangled up in red: intertwining of the heme and iron-sulfur cluster biogenesis pathways. Cell Metab. 2009;10(2):80–1.
Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007;21(1):1–20.
Wu J, Wang J, Zeng X, Chen Y, Xia J, Wang S, et al. Protein phosphatase 2A regulatory subunit B56β modulates erythroid differentiation. Biochem Biophys Res Commun. 2016;478(3):1179–84.
Thom CS, Traxler EA, Khandros E, Nickas JM, Zhou OY, Lazarus JE, et al. Trim58 degrades dynein and regulates terminal erythropoiesis. Dev Cell. 2014;30(6):688–700.
Tanno T, Miller JL. Iron loading and overloading due to ineffective erythropoiesis. Adv Hematol. 2010;2010:358283.
Soler Artigas M, Wain LV, Repapi E, Obeidat M, Sayers I, Burton PR, et al. Effect of five genetic variants associated with lung function on the risk of chronic obstructive lung disease, and their joint effects on lung function. Am J Respir Crit Care Med. 2011;184(7):786–95.
Zhang BL, Xu RL, Qin YW, Zheng X, Wu H, You XH, et al. Potential candidate genes for alveolar hypoxia identified by transcriptome network analysis. Medicina (Kaunas). 2012;48(11):572–80.
Kent BD, Mitchell PD, McNicholas WT. Hypoxemia in patients with COPD: cause, effects, and disease progression. Int J Chron Obstruct Pulmon Dis. 2011;6:199–208.
Zhao B, Keerthivasan G, Mei Y, Yang J, McElherne J, Wong P, et al. Targeted shRNA screening identified critical roles of pleckstrin-2 in erythropoiesis. Haematologica. 2014;99(7):1157–67.
Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A. 2001;98(15):8780–5.
Bellet MM, Piobbico D, Bartoli D, Castelli M, Pieroni S, Brunacci C, et al. NEDD4 controls the expression of GUCD1, a protein upregulated in proliferating liver cells. Cell Cycle. 2014;13(12):1902–11.
Schols AM, Gosker HR. The pathophysiology of cachexia in chronic obstructive pulmonary disease. Curr Opin Support Palliat Care. 2009;3(4):282–7.
Acknowledgements
Not Applicable.
Funding
Funding was provided by the National Institutes for Health grant R00HL121087 and the Parker B. Francis Fellowship Program, R01HL130512, R01HL125583. ECLIPSE was funded by GSK. The COPDGene study is supported by NIH grants U01 HL089856 and U01 HL089897 and is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, Novartis, GlaxoSmithKline, Pfizer, Siemens and Sunovion. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agents.
Author information
Authors and Affiliations
Contributions
Conception and design of this study and creation, revision, and final approval of this manuscript: A.C.W., P.L.K., and M.-L.N.M.; analysis and interpretation: A.C.W., P.L.K., H.T., I.A., J.D.M. E.F.M.W., R.C., S.I.R., D.A.L., B.R.C., A.A., R.T.-S., M.T.D., S.B., V.J.T., C.P.H., J.M.W., P.J.C., and E.K.S. and M.-L.N.M.; data acquisition: E.F.M.W., R.C., S.I.R., D.A.L., B.R.C., A.A., R.T.-S., M.T.D., C.P.H., P.J.C., G.W., and E.K.S.; drafting the manuscript for important intellectual content: A.C.W. and M.-L.N.M. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Institutional Review Board approval for all analyses was obtained from the University of Alabama at Birmingham and have been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All subjects included in this analysis provided written informed consent prior to their inclusion in the respective studies mentioned below.
Consent for publication
Not Applicable.
Competing interests
ACW, PLK, SL, MMP, IA, HKT, VJT, GW, MW, AA, SR, and RC report no conflicts of interest. JM reports grants from NIH NHLBI, during the conduct of the study. EW reports board membership for Nycomed and Boehringer, grants from AstraZeneca and GlaxoSmithKline, and payment for lectures including service on speakers’ bureaus from GlaxoSmithKline, Novartis, and Chiesi. DL has received grant support, honoraria and consultancy fees from GSK. SPB is supported by NIH grant K23HL133438. He has served on advisory boards for Sunovion and GlaxoSmithKline. CH reports grants from National Institutes of Health, during the conduct of the study; personal fees from AstraZeneca, personal fees from Concert Pharmaceuticals, personal fees from 23andMe, grants from Novartis, grants from Boehringer-Ingelheim, outside the submitted work. EKS received grants and travel support from GlaxoSmithKline within the past three years. MLM reports grants from NIH, other from Parker B. Francis Foundation, during the conduct of the study; personal fees from Pfizer, outside the submitted work. RTS reports personal fees from GlaxoSmithKline, during the conduct of the study. MTD reports grants from NIH, American Lung Association, Department of Defense, and the Department of Veterans Affairs; personal fees from AstraZeneca, Boehringer Ingelheim, PneumRx/BTG, Mereo, Quark Pharmaceuticals and GSK, and contracted clinical trial support from Boehringer Ingelheim, Novartis, AstraZeneca, Yungjin, PneumRx/BTG, Pulmonx, Boston Scientific, Gala, Nuvaira, and GSK. PJC has received research support and consulting fees from GSK and Novartis. JMW reports grants from NIH/NHLBI, during the conduct of the study; grants from NIH/NCATS, grants from Bayer, grants and other from GSK, other from Boehringer Ingelheim, grants and other from Mereo BioPharma, other from PRA, outside the submitted work; .
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
ECLIPSE Sample Demographics. Descriptive values for COPD patients (N = 114) in ECLIPSE with gene expression data by Cachectic (N = 9) and Non-Cachectic (N = 105) co-morbid status. Unless otherwise noted values denote mean (SD). Table S2. Gene set enrichment analysis of 23 significantly (FDR p-value < 0.05) differentially expressed between cachectic and non-cachectic COPD patients in COPDGene.
Additional file 2: Figure S1.
Principle component analysis on SV residualized expression data of (N = 400) COPDGene subjects with COPD. Principal component 1 (PC1) is presented on the x-axis and the second principle component on the y-axis (PC2) after adjustment for significant surrogate variables (N = 27). Numbers in parentheses denotes the proportion of variance captured by each component. Red dots indicate COPD subjects with cachexia. Blue dots indicate COPD subjects without cachexia.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Wilson, A.C., Kumar, P.L., Lee, S. et al. Heme metabolism genes Downregulated in COPD Cachexia. Respir Res 21, 100 (2020). https://doi.org/10.1186/s12931-020-01336-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-020-01336-w