
Abstract
Background
We conducted a genome-wide association study (GWAS) of subclinical interstitial lung disease (ILD), defined as high attenuation areas (HAA) on CT, in the population-based Multi-Ethnic Study of Atherosclerosis Study.
Methods
We measured the percentage of high attenuation areas (HAA) in the lung fields on cardiac CT scan defined as voxels with CT attenuation values between -600 and -250 HU. Genetic analyses were performed in MESA combined across race/ethnic groups: non-Hispanic White (n = 2,434), African American (n = 2,470), Hispanic (n = 2,065) and Chinese (n = 702), as well as stratified by race/ethnicity.
Results
Among 7,671 participants, regions at genome-wide significance were identified for basilar peel-core ratio of HAA in FLJ35282 downstream of ANRIL (rs7852363, P = 2.1x10−9) and within introns of SNAI3-AS1 (rs140142658, P = 9.6x10−9) and D21S2088E (rs3079677, P = 2.3x10−8). Within race/ethnic groups, 18 additional loci were identified at genome-wide significance, including genes related to development (FOXP4), cell adhesion (ALCAM) and glycosylation (GNPDA2, GYPC, GFPT1 and FUT10). Among these loci, SNP rs6844387 near GNPDA2 demonstrated nominal evidence of replication in analysis of n = 1,959 participants from the Framingham Heart Study (P = 0.029). FOXP4 region SNP rs2894439 demonstrated evidence of validation in analysis of n = 228 White ILD cases from the Columbia ILD Study compared to race/ethnicity-matched controls from MESA (one-sided P = 0.007). In lung tissue from 15 adults with idiopathic pulmonary fibrosis compared to 15 adults without lung disease. ANRIL (P = 0.001), ALCAM (P = 0.03) and FOXP4 (P = 0.046) were differentially expressed.
Conclusions
Our results suggest novel roles for protein glycosylation and cell cycle disinhibition by long non-coding RNA in the pathogenesis of ILD.
Similar content being viewed by others


Background
The interstitial lung diseases (ILDs) are a family of non-infectious, non-malignant lung diseases characterized by alveolar injury, inflammation, and fibrosis. Idiopathic pulmonary fibrosis (IPF), the most common idiopathic ILD, has a median survival time of 3.8 years and affects nearly 1 out of 200 older adults in the United States [1]. Previous genome-wide association studies (GWAS) for IPF have demonstrated the utility of genetic approaches for discovering novel biological pathways involved in the pathogenesis of ILD in studies of up to ~1,600 cases and ~4,700 controls [2, 3]. Variation in the promoter of MUC5B was identified as a risk factor for clinically evident sporadic and familial ILD [4] and is associated with the presence of interstitial lung abnormalities (ILAs), a qualitative subclinical ILD phenotype, in the Framingham Heart Study (FHS) [5]. Other genes implicated in the development of ILD include SFTPA2, SFTPC, ELMOD2, TERT, TERC, FAM13A, DSP, OBFC1, DPP9, TOLLIP, ATP11A, and SPPL2C [2, 3].
The study of early, subclinical disease before advanced parenchymal changes have occurred could lead to the identification of novel biological pathways involved in the pathogenesis of ILD at a stage more amenable for intervention. High attenuation areas (HAA), defined as the percentage of lung voxels in the range of -600 – −250 Hounsfield units, correspond to the CT attenuation values observed in areas visually described as having ground-glass and reticular attenuation [6]. In the Multi-Ethnic Study of Atherosclerosis (MESA), we have shown that greater HAA is associated with cigarette smoking, lower forced vital capacity, reduced exercise capacity, higher serum levels of matrix metalloproteinase-7 (MMP-7) and interleukin-6, a higher prevalence of interstitial lung abnormalities (ILAs) at 9.5 years follow-up, and a higher risk of death [6, 7].
Under the hypothesis that high throughput genetic association analysis could identify novel pathophysiologic pathways to explain the occurrence of HAA and ILD, we conducted a GWAS for percent HAA on cardiac CT in the MESA Study. We further sought replication of identified SNPs for percent HAA in participants of European ancestry from the FHS, validation in analysis of ILD cases from the Columbia ILD Study compared to matched controls selected from among the MESA participants, and performed differential gene expression analysis for comparing mRNA from lung tissue of IPF cases versus controls.
Methods
Study participants
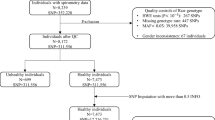
MESA is a population-based longitudinal study of subclinical cardiovascular disease [8]. Between 2000 and 2002, MESA recruited 6,814 men and women 45–84 years of age from six US sites who were free of clinical cardiovascular disease. The MESA Family Study recruited an additional 1,595 African-American and Hispanic family members 45–84 years of age specifically for genetic analysis, and the MESA Air Pollution Study recruited an additional 257 participants [9]. Participants who did not consent to genetic analyses or who had no usable genetic material were excluded, resulting in a combined sample of 7,671 participants comprising the MESA SNP Health Association Resource (SHARe) sample, which has been previously described [10].
We sought replication of selected SNPs from the race/ethnic specific GWAS of MESA Whites in the Framingham Heart Study (FHS), an independent population-based cohort. See Online Supplement for details.
Percent high attenuation areas
The MESA Lung Fibrosis Study was developed to determine the percent HAA for all participants on the lung fields of cardiac CT scans, which were acquired under a standardized protocol [6, 11]. For discovery analyses in MESA as well as replication in FHS, percent HAA was defined at lung regions between -600 and -250 Hounsfield Units (HU), with basilar percent HAA quantified as the percent HAA in the caudal 1/3rd of the imaged lung. In MESA, basilar peel-core ratio of HAA (henceforth termed “basilar peel-core ratio”) computed as the percent HAA in the peel region (outer 20 mm) divided by that in the core region for the caudal 1/3rd of the imaged lung.
Genotyping
Genome-wide genotyping was performed for MESA and FHS participants followed by imputation the 1000 Genomes reference panel [12]. Additional details are provided in the Online Supplement.
Genetic association analysis
Within the MESA Lung Fibrosis study, we completed GWAS analyses of percent HAA, basilar percent HAA, and basilar peel-core ratio. Race/ethnic-specific analyses were performed in linear regression models with adjustment for age, sex, study site, principal components (PCs) of ancestry, CT scanner, tube current, breath artifacts, height, weight, cigarettes per day (for current smokers only) and pack-years of smoking. We did not adjust for dichotomous smoking exposures (current/former/never smoking) because this information was already captured by the continuous measures of smoking exposure. Results were combined by fixed-effect meta-analyses across all four MESA race/ethnic groups.
We examined genomic control values of all GWAS for evidence of residual population stratification, undetected family structure, or other sources of inflation in type I error. Single SNP association results that attained a threshold of P < 5.0 x 10−8 were considered genome-wide significant. Those SNPs demonstrating genome-wide significant association in discovery GWAS of percent HAA and basilar percent HAA in MESA were examined for evidence of replication with adjustment for the same covariates in FHS. Additional details provided in the Online Supplement.
Validation with ILD cases
Three hundred sixteen ILD cases (108 of these were IPF confirmed) were recruited between 2007 and 2011 through the Columbia ILD Study, an NIH-funded prospective cohort study at Columbia University Medical Center. For selected SNPs, we performed genetic analysis with these ILD cases compared to MESA controls matched on race/ethnicity. See Online Supplement for details.
Gene expression analyses
We measured mRNA expression of four of the genes we identified (ALCAM, FOXP4, CDKN2B, and ANRIL, along with the reference gene GAPDH) in OCT-embedded fresh frozen lung tissue obtained from 15 adults with IPF and a histologic usual interstitial pneumonia pattern (UIP) and 15 adults without lung disease stored in the Columbia University Pathology Tissue Bank. See Online Supplement for details.
Results
Characteristics of the MESA lung fibrosis participants
MESA Lung Fibrosis participants [10] represent a relatively healthy cohort ages 39–91 years, 54% of whom were female, with race/ethnic distribution consisting of 32% non-Hispanic White (henceforth termed “White”), 32% African American, 27% Hispanic, and 9% Chinese. Smoking history varied considerably across race/ethnic groups: 56% of Whites, 53% of African Americans, 45% of Hispanics, and 25% of Chinese participants had ever smoked.
The median percent HAA ranged from 3.9% in Whites to 4.5% in Hispanic and Chinese, and median basilar peel-core ratio ranged from 0.89 in Chinese to 0.96 in Whites. Percent HAA and basilar peel-core ratio were relatively distinct from one another, with correlations for ranging from 0.01 to 0.17 within race/ethnic groups (Table 1).
GWAS of HAA phenotypes across race/ethnic groups
In combined analysis across race/ethnic groups, we identified three SNPs at genome-wide significance for basilar peel-core ratio (Table 2, Fig. 1, Additional file 1: Figures S1-S2). The top SNP for the first locus, rs7852363 (P = 2.1x10−9), lies within the long non-coding RNA gene FLJ35282, and about 670 kb downstream of the 9p21 locus near the long non-coding RNA ANRIL, widely reported in association with coronary disease risk [13]. The top associated SNP for the second locus, rs140142658 (P = 9.6x10−9), lies within the first intron of the antisense RNA gene SNAI3-AS1. The top associated variant for the third locus, rs3079677 (P = 2.3x10−8), is a deletion within the last intron of the long non-coding RNA gene D21S2088E. For all three of these loci, the effect allele is associated with increased basilar peel-core ratio across each of the MESA race/ethnic groups contributing to the combined result (Additional file 1: Table S1).

Regional association plots for statistically significant GWAS regions identified in association with basilar peel-core ratio based on meta-analysis to combine results across race/ethnic groups in MESA. a Chromosome 9 region including FLJ35282, b Chromosome 16 region at SNAI3-AS1 and c Chromosome 21 region at D21S2088E. All plots are generated using the LocusZoom tool [36] with 1000 Genomes EUR (European ancestry) reference panel used for calculating linkage disequilibrium (LD). The x-axis shows position on the indicated chromosome in megabases (Mb), while the y-axis displays strength of association for each SNP as –log [10] p-value (left) and recombination rate at each genomic position (right). Genotyped SNPs are marked as crosses and imputed SNPs are shown as circles. The SNP indicated in purple is the index SNP, and other SNPs are color coded according to their linkage disequilibrium (R-squared) with the index SNP. LD for (c) is shown with respect to the SNP rs1504542 because LD information was not available for rs3079677 in the selected reference panel
GWAS of HAA phenotypes within race/ethnic groups
Among Whites, four loci in or near the genes GNPDA2, FOXP4, ZNF664-FAM101A and DAAM1 demonstrated genome-wide significant association with percent HAA (Additional file 1: Figure S3), while three additional loci in or near UBE2U, GYPC, and ALCAM showed genome-wide significant association with basilar HAA (Additional file 1: Figure S4). For basilar peel-core ratio, we also identified genome-wide significance for four loci at SLC45A1, FAM69A, GFPT1, and SEH1L (Additional file 1: Figure S5). The identified SNPs were rare/infrequent in Whites with minor allele frequency (MAF) ranging from 0.006 to 0.026 and the minor alleles were all associated with increased HAA, basilar HAA or basilar peel-core ratio (Table 2). Common themes and putative functions of genes identified in Whites include genes that act on glycoproteins and glycosylation (GNPDA2, GYPC, GFPT1), genes important in development (FOXP4, DAAM1) and cell adhesion (DAAM1, ALCAM).
Among African Americans, we identified three loci in or near the genes STK38, FUT10 and SAMD4A associated with basilar HAA and two loci intronic to the genes NCOA2 and SLC25A21 associated with basilar peel-core ratio.
Among Chinese, we identified a locus at genome-wide significance in PRKP for percent HAA and near SORCS3 for basilar percent HAA.
While we noted overlap in the putative function of genes identified in the race/ethnic specific GWAS of HAA, specific variants identified in race/ethnic specific analyses did not generally exhibit consistent effects across race/ethnic groups (Additional file 1: Table S1-S3). SNPs that did show consistent directions of effect across race/ethnic groups include rs190432524 at GFPT2 for basilar peel-core ratio (Additional file 1: Table S1), and rs6844387 at GNPDA2 for percent HAA (Additional file 1: Table S2). The results identified for basilar percent HAA appeared to be mostly race/ethnic specific, without shared evidence across groups (Additional file 1: Table S3). Sensitivity analysis demonstrated the main SNP associations reported in Table 2 were largely unchanged after additional adjustment for BMI, waist circumference and diabetes status (Additional file 1: Table S4). Many of the main SNP associations were also robust to covariate adjustment for percent emphysema (Additional file 1: Table S5). However, some SNPs showed attenuated evidence for association after adjustment for percent emphysema resulting from residual association with percent emphysema (Additional file 1: Table S6). In particular, the FOXP4 SNP rs2894439 demonstrated the strongest evidence of with percent emphysema (P = 4.1x10−6), although the strength of association with percent emphysema did not reach that seen for percent HAA in primary analysis (Table 2, P = 3.5x10−8).
Replication in the Framingham heart study
We examined evidence of replication in N = 1959 participants from the Framingham Heart Study (FHS, Additional file 1: Table S7) for the seven SNPs identified in the GWAS of percent HAA and basilar HAA in MESA Whites (shown in Table 2). One SNP rs6844387 near GNPDA2 demonstrated nominal significance (Additional file 1: Table S8, P = 0.029) in this group with the same direction of effect as observed in MESA (Table 2). Meta-analysis across MESA Whites and FHS yielded an attenuated effect size reflecting the smaller effect observed in FHS (combined Beta = 0.419, P = 2.3x10−11).
Validation with ILD cases
In race/ethnic specific analysis of Whites, we examined evidence of association for 13 SNPs identified in the GWAS of percent HAA, basilar percent HAA, or basilar peel-core ratio for MESA Whites or in meta-analysis across race/ethnic groups (Table 2). SNP rs2894439 near FOXP4 demonstrated nominally significant association (Additional file 1: Table S9-S10, one-sided P = 0.007) in Whites, with increased frequency in Columbia ILD cases (MAF = 0.048) compared to matched controls free of ILD in MESA (MAF = 0.021). We did not observe any SNPs reaching nominal significance in the combined analysis across race/ethnic groups.
Examination of SNPs reported in previous GWAS of IPF
We evaluated MESA GWAS results for those SNPs that have been reported in previously published GWAS of IPF [2,3,4, 14], and identified a total of 33 SNPs of interest as shown in Additional file 1: Table S11. After correction for multiple comparisons (P < 0.05/33 comparisons = 0.0015), two intronic SNPs for TOLLIP exhibited significant associations with increased percent HAA (Additional file 1: Table S12, rs5743894, P = 0.0009) and basilar percent HAA (Additional file 1: Table S13, rs5743894, P = 0.0007) in African Americans.
Gene expression
We selected 4 genes (ANRIL, ALCAM, FOXP4, and CDKN2B) for gene expression analyses. We identified a 3-fold increase in ANRIL mRNA expression in IPF lung compared to controls (Fig. 2; mean fold change 3.1, 95% CI 1.7 – 5.6, P = 0.001). ANRIL mRNA localized to airway epithelium in both UIP and normal lung, with no expression in fibroblastic foci (Fig. 3). Expression of ALCAM (mean fold change 0.58, 95% CI 0.40 – 0.85, P =0.03) and FOXP4 (mean fold change 0.70, 95% CI 0.50 – 0.98, P = 0.046) were decreased in IPF lung compared to controls (Fig. 2). There was a non-significant trend toward lower CDKN2B expression in IPF lung compared to normal lung (mean fold change 0.56, 95% CI 0.33 – 0.94, P = 0.07).

mRNA expression in whole lung tissue for ANRIL, CDKN2B, ALCAM, and FOXP4 from 15 cases with IPF (red) and 15 controls (white) using quantitative real-time PCR normalized to expression of the reference gene GADPH. Results are presented as boxplots of the fold difference in expression in each IPF case or control normalized to the mean expression value among controls. Within each boxplot, the thick horizontal line represents the median fold difference, the ends of the boxplots are placed at the 25th and 75th percentiles (interquartile range), the whiskers extend to 1.5 x the interquartile range, and outliers are represented by open circles. Mean fold change (MFC) was calculated using the ΔΔCt method. P values are from Wilcoxon rank sum tests. The horizontal line at a value of 1 is the mean normalized expression value among controls

Expression of ANRIL (a long non-coding RNA) in UIP lung tissue. H&E stained tissue (left: panels a and c); in situ hybridization of ANRIL transcripts (right: panels b and d) for airway epithelium (top: panels a and b) versus fibroblastic foci (bottom: panels c and d). Note the pink dots (ANRIL transcripts) in airway epithelium (b) but not in fibroblastic foci (d). The photomicrograph is presented at x150 magnification
Discussion
In this multi-ethnic GWAS of ~8,000 individuals, we identified associations between HAA phenotypes and the 9p21 region and two other loci also near other RNA genes.
In race/ethnic-specific analyses, we identified a number of genes related to obesity (GNPDA2, ZNF664-FAM101A, PFKP, SAMD4A), glycosylation (GYPC, FUT10), and carbohydrate metabolism (GNPDA2, PFKP, SLC45A). We also identified novel associations with genes that code for a transcription factor (FOXP4) that validated in clinical ILD cases, a cell adhesion molecule (ALCAM) expressed in the pulmonary microvascular endothelium, a protein involved in reorganization of the actin cytoskeleton (DAAM1) that may play a role in pulmonary vascular remodeling, and a (STK38) MAP kinase inhibiting protein that protects against acute lung injury.
Our major finding is that ANRIL expression is higher in IPF lung compared to normal lung (Additional file 1: Table S14). This discovery came as a result of follow-up on our GWAS finding that HAA, a putative measure of subclinical ILD, is associated with genetic variation at rs7852363, a SNP that sits within the long non-coding RNA FLJ35282 downstream of the CDKN2A/CDKN2B/ANRIL locus. The latter has been strongly linked to cardiovascular disease [13], diabetes [15], and cancer [16]. Evidence suggests that much of the phenotypic variation linked to 9p21 may be explained by genotypic variation in ANRIL [17], a long non-coding RNA (lncRNA) gene that might promote cis-acting epigenetic silencing of the 9p21 region by promoting recruitment of polycomb repressive complexes [18], leading to decreased expression of CDKN2A and CDKN2B. A recent study found that CDKN2B is highly methylated in IPF fibroblasts, possibly contributing to increased cyclin-dependent kinase activity and fibroblast proliferation in IPF [19].
Our findings of increased ANRIL expression and reduced CDKN2B expression in IPF lung support a possible role for disinhibition of cyclin-dependent kinases in IPF progression. The localization of ANRIL expression to the airway epithelium, however, suggests that if ANRIL plays a role in IPF pathogenesis, it might contribute to an abnormal small airway epithelial response to injury rather than excess fibroblast proliferation. We did not seek validation of rs7852363 in the Framingham Heart Study, since the phenotype demonstrating an association with this locus (basilar HAA peel-to-core ratio) was not available. Although we were unable to demonstrate an association between rs7852363 and clinical ILD case status, the small number of clinical cases, combined with our above findings, suggest that additional work in this area is needed.
We found that the FOXP4 minor allele was present in 4.8% of ILD cases compared to 2.1% of controls, and FOXP4 also demonstrated reduced expression in lung tissue from IPF cases compared to controls. FOXP4 encodes a transcription factor (forkhead box P4) expressed in proximal and distal airway epithelium as well as in subepithelial mesenchyme during lung development [20]. FOXP4 is also expressed in CD4+ and CD8+ T cells and appears to contribute to cytokine production and memory T cell responses [21], and a recent study reported FOXP4 as an important regulator of non-small cell lung cancer cells [22]. The specific role of this locus in disease pathogenesis requires further elucidation.
In our replication analysis for percent HAA, only one SNP near the GNPDA2 demonstrated nominal evidence of association in European ancestry participants from FHS. Given the number of SNPs considered in the replication analysis, nominally significant evidence of association does not pass correction for multiple comparisons, but perhaps offers a suggestion of association. GNPDA2 catalyzes conversion of D-glucosamine-6-phosphate into D-fructose-6-phosphate, representing a possible relationship with catabolism of N-linked carbohydrates in glycoproteins, similar to GYPC, GFPT1 and FUT10 also reported in our current GWAS of HAA in MESA. Glycosylation plays a key role in quality control of glycoproteins [23]. In the lung, mucins are heavily glycosylated proteins produced in the epithelium and have been among the most widely replicated association for ILD at MUC5B [2, 4, 5]. Altered glycosylation of mucin and/or extracellular matrix proteins in the lung could impair remodeling responses to injury and promote interstitial lung disease [24]. Our novel findings of associations between subclinical ILD and variation at loci responsible for glycosylation suggest that investigation of the role of protein glycosylation in early lung remodeling may provide insights into pathogenesis of ILD.
In analysis of 33 SNPs reported previous GWAS of IPF, we identified association of two TOLLIP SNPs with percent HAA traits in MESA African Americans. Combined with the previous association of the same TOLLIP SNPs with IPF [3], our identification of TOLLIP SNPs associated with percent HAA suggests these SNPs play a role in subclinical disease progressing toward clinical development of pulmonary fibrosis. TOLLIP (also known as Toll interacting protein) has been shown to regulate inflammatory cytokine production in response to interleukin-1 [25], identifying inflammation as a possible mechanism underlying the role of TOLLIP in development of IPF.
While we were unable to validate other SNPs that we identified in our GWAS, literature evidence provides some support for a role in ILD pathogenesis for three genes located in or near SNPs identified by our GWAS. Activated leukocyte cell adhesion molecule (CD166; ALCAM) is an immunoglobulin-family receptor expressed on activated leukocytes [26] and in some cancers. We found that ALCAM is under-expressed in IPF lung. A recent exome sequence analysis of severe early onset COPD exhibited an increased burden of rare deleterious ALCAM variants in cases versus controls [27]. ALCAM may play a signaling role in extracellular matrix remodeling [28] and is expressed in the pulmonary microvascular endothelium [29]. DAAM1 encodes for disheveled association activator of morphogenesis 1, a protein involved in actin assembly [30] and endothelial cell proliferation [31]. DAAM1 is overexpressed in pulmonary arteries in idiopathic pulmonary arterial hypertension [32]. STK38 encodes for serine-threonine kinase 38, an inhibitor of mitogen-activated protein (MAP) kinase 1 signaling [33] has been linked to protection from nickel-induced acute lung injury [34].
Limitations of the study include a modest sample size for GWAS, particularly among Chinese, such that reported associations should be interpreted with caution. However, the present study is the first and largest GWAS of subclinical ILD phenotypes to date. In addition, data available for replication was limited to European ancestry participants from the Framingham Heart Study who were assessed for percent HAA and basilar percent HAA, but not for basilar peel-core ratio. Accordingly, we could not meaningfully pursue replication of genetic loci identified in non-European ancestry groups from MESA. Further, while the MESA participants were genotyped for GWAS at ~900,000 SNPs using the Affymetrix 6.0 array which was designed to capture common variation across Europeans, East Asians and West Africans [35], we expect that we would have had limited coverage of common variants with MAF less than 0.1 even after incorporating imputation to the 1000 Genomes reference panel. Therefore, despite the fact that the genetic loci reported from our GWAS were subject to strict and systematic filters on imputation quality and minor allele counts, we recognize that many of our reported loci reflect infrequent variants that should be viewed with caution, particularly in the absence of replication and lack of consistent effects across race/ethnic groups. Given that we have focused on a population-based cohort, our study may have reduced power to detect infrequent risk variants seen predominantly in ILD cases. We expect our use of quantitative measures to define subclinical ILD provided us with greater power to identify infrequent variants that confer protection against ILD. Taken together, the limitations of our study underscore the fact that the SNPs and genes implicated by our current work will require additional confirmation through replication and validation in larger sample sizes.
Conclusions
In summary, we report the first genome-wide association study to probe the genetics of HAA in a general population sample, and the novel finding that ANRIL, a long non-coding RNA that promotes loss of cell cycle regulation, is overexpressed in IPF lung. We also report a novel FOXP4 region SNP that was validated in genetic analysis of ILD cases versus controls, and further demonstrate reduced expression of FOXP4 in lung tissue from IPF cases compared to controls. Our results suggest morphologic assessment of the lung on CT can build substantially on our knowledge of ILD pathobiology and that disinhibition of cyclin-dependent kinases and abnormalities in protein glycosylation may contribute to ILD risk.
Abbreviations
- CT:
-
Computed tomography
- GWAS:
-
Genome-wide association study
- HAA:
-
High attenuation area
- HU:
-
Hounsfield unit
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- MESA:
-
The Multi-Ethnic Study of Atherosclerosis
- SNP:
-
Single nucleotide polymorphism
References
Raghu G, Lynch D, Godwin JD, Webb R, Colby TV, Leslie KO, et al. Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: secondary analysis of a randomised, controlled trial. Lancet Respir Med. 2014;2:277–84.
Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–20.
Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–17.
Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011;364:1503–12.
Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med. 2013;368:2192–200.
Lederer DJ, Enright PL, Kawut SM, Hoffman EA, Hunninghake G, van Beek EJ, et al. Cigarette smoking is associated with subclinical parenchymal lung disease: the multi-ethnic study of atherosclerosis (MESA)-lung study. Am J Respir Crit Care Med. 2009;180:407–14.
Podolanczuk AJ, Oelsner EC, Barr RG, Hoffman EA, Armstrong HF, Austin JH, et al. High attenuation areas on chest computed tomography in community-dwelling adults: the MESA study. Eur Respir J. 2016;48:1442–52.
Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–81.
Kaufman JD, Adar SD, Allen RW, Barr RG, Budoff MJ, Burke GL, et al. Prospective study of particulate air pollution exposures, subclinical atherosclerosis, and clinical cardiovascular disease: the multi-ethnic study of atherosclerosis and air pollution (MESA Air). Am J Epidemiol. 2012;176:825–37.
Manichaikul A, Hoffman EA, Smolonska J, Gao W, Cho MH, Baumhauer H, et al. Genome-wide study of percent emphysema on computed tomography in the general population. The multi-ethnic study of atherosclerosis lung/SNP health association resource study. Am J Respir Crit Care Med. 2014;189:408–18.
Carr JJ, Nelson JC, Wong ND, McNitt-Gray M, Arad Y, Jacobs Jr DR, et al. Calcified coronary artery plaque measurement with cardiac CT in population-based studies: standardized protocol of multi-ethnic study of atherosclerosis (MESA) and coronary artery risk development in young adults (CARDIA) study. Radiology. 2005;234:35–43.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65.
O’Donnell CJ, Kavousi M, Smith AV, Kardia SL, Feitosa MF, Hwang SJ, et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation. 2011;124:2855–64.
Mushiroda T, Wattanapokayakit S, Takahashi A, Nukiwa T, Kudoh S, Ogura T, et al. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J Med Genet. 2008;45:654–6.
Broadbent HM, Peden JF, Lorkowski S, Goel A, Ongen H, Green F, et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet. 2008;17:806–14.
Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–6.
Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. 2010;6:e1000899.
Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–74.
Huang SK, Scruggs AM, McEachin RC, White ES, Peters-Golden M. Lung fibroblasts from patients with idiopathic pulmonary fibrosis exhibit genome-wide differences in DNA methylation compared to fibroblasts from nonfibrotic lung. PLoS One. 2014;9:e107055.
Lu MM, Li S, Yang H, Morrisey EE. Foxp4: a novel member of the Foxp subfamily of winged-helix genes co-expressed with Foxp1 and Foxp2 in pulmonary and gut tissues. Mech Dev. 2002;119 Suppl 1:S197–202.
Wiehagen KR, Corbo-Rodgers E, Li S, Staub ES, Hunter CA, Morrisey EE, et al. Foxp4 is dispensable for T cell development, but required for robust recall responses. PLoS One. 2012;7:e42273.
Yang T, Li H, Thakur A, Chen T, Xue J, Li D, et al. FOXP4 modulates tumor growth and independently associates with miR-138 in non-small cell lung cancer cells. Tumour Biol. 2015;36:8185–91.
Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13:448–62.
Nicolaou N, Margadant C, Kevelam SH, Lilien MR, Oosterveld MJ, Kreft M, et al. Gain of glycosylation in integrin alpha3 causes lung disease and nephrotic syndrome. J Clin Invest. 2012;122:4375–87.
Didierlaurent A, Brissoni B, Velin D, Aebi N, Tardivel A, Kaslin E, et al. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Mol Cell Biol. 2006;26:735–42.
Bowen MA, Patel DD, Li X, Modrell B, Malacko AR, Wang WC, et al. Cloning, mapping, and characterization of activated leukocyte-cell adhesion molecule (ALCAM), a CD6 ligand. J Exp Med. 1995;181:2213–20.
Qiao D, Lange C, Beaty TH, Crapo JD, Barnes KC, Bamshad M, et al. Exome sequencing analysis in severe, early-onset chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2016;193:1353–63.
Lunter PC, van Kilsdonk JW, van Beek H, Cornelissen IM, Bergers M, Willems PH, et al. Activated leukocyte cell adhesion molecule (ALCAM/CD166/MEMD), a novel actor in invasive growth, controls matrix metalloproteinase activity. Cancer Res. 2005;65:8801–8.
Ofori-Acquah SF, King J, Voelkel N, Schaphorst KL, Stevens T. Heterogeneity of barrier function in the lung reflects diversity in endothelial cell junctions. Microvasc Res. 2008;75:391–402.
Bradley MN, Hong C, Chen M, Joseph SB, Wilpitz DC, Wang X, et al. Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. J Clin Invest. 2007;117:2337–46.
Ju R, Cirone P, Lin S, Griesbach H, Slusarski DC, Crews CM. Activation of the planar cell polarity formin DAAM1 leads to inhibition of endothelial cell proliferation, migration, and angiogenesis. Proc Natl Acad Sci U S A. 2010;107:6906–11.
Laumanns IP, Fink L, Wilhelm J, Wolff JC, Mitnacht-Kraus R, Graef-Hoechst S, et al. The noncanonical WNT pathway is operative in idiopathic pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2009;40:683–91.
Enomoto A, Kido N, Ito M, Morita A, Matsumoto Y, Takamatsu N, et al. Negative regulation of MEKK1/2 signaling by serine-threonine kinase 38 (STK38). Oncogene. 2008;27:1930–8.
Mongan M, Tan Z, Chen L, Peng Z, Dietsch M, Su B, et al. Mitogen-activated protein kinase kinase kinase 1 protects against nickel-induced acute lung injury. Toxicol Sci. 2008;104:405–11.
Hoffmann TJ, Kvale MN, Hesselson SE, Zhan Y, Aquino C, Cao Y, et al. Next generation genome-wide association tool: design and coverage of a high-throughput European-optimized SNP array. Genomics. 2011;98:79–89.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7.
Acknowledgments
The authors thank the participants, investigators and study staff of MESA, FHS and the Columbia IPF Study.
Funding
MESA and the MESA SHARe project are conducted and supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with MESA investigators. Support for MESA is provided by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169, UL1-TR-001079, UL1-TR-000040, and DK-063491. MESA Family is conducted and supported by the NHLBI in collaboration with MESA investigators. Support is provided by grants and contracts R01HL071051, R01HL071205, R01HL071250, R01HL071251, R01HL071258, R01HL071259, by the National Center for Research Resources, Grant UL1RR033176, and the National Center for Advancing Translational Sciences, Grant UL1TR000124. MESA Air is conducted and supported by the United States Environmental Protection Agency (EPA) in collaboration with MESA Air investigators, with support provided by grant RD831697. Funding for SHARe genotyping was provided by NHLBI Contract N02-HL-64278. Genotyping was performed at Affymetrix (Santa Clara, California, USA) and the Broad Institute of Harvard and MIT (Boston, Massachusetts, USA) using the Affymetrix Genome-Wide Human SNP Array 6.0. The MESA Lung and MESA COPD Studies are funded by NIH grants R01HL077612 and R01HL093081. The MESA Lung Fibrosis Study was funded by NIH R01HL103676, K24HL131937, and by a grant from the Pulmonary Fibrosis Foundation. The MESA Lung/SHARe Study was funded by NIH grant RC1HL100543. This study was supported by NIH K23HL086714, KL2 TR000081, UL1 TR000040 and R01HL131565.
The Framingham Heart Study was supported by the National Heart, Lung, and Blood Institute’s Framingham Heart Study (contract number N01-HC-25195) and its contract with Affymetrix, Inc., for genotyping services (contract number N02-HL-6-4278).
Availability of data and materials
MESA SHARe genotype and phenotype data used for the current analyses are publicly available on dbGaP (https://dbgap.ncbi.nlm.nih.gov) under study accession number phs000209.v7.p2.
Framingham SHARe genotype and phenotype data used for replication analysis are publicly available on dbGaP (https://dbgap.ncbi.nlm.nih.gov) under study accession number phs000007.v28.p10.
Authors’ contributions
AM, JD, GR, GRW, DR, SMK, GTO, SSR, RGB, and DJL contributed to study design. AM, JD, EAH, JDK, KDHS, SMK, GMH, GRW, GTO, RGB, and DJL performed phenotype data acquisition and quality control. AM, XQW, LS, JD, SO-G, EAF, GTO, and SSR carried out genotype data acquisition and quality control. LS and ACB performed tissue acquisition and staining, pathology review and interpretation, and gene expression analysis. AM, XQW, JD, JNN, LS, SSR, RGB, and DJL performed data analysis. All authors read and approved the final manuscript.
Competing interests
EAH is a founder and shareholder in VIDA Diagnostics. SMK reports non-financial support from the American Thoracic Society, personal fees from the European Respiratory Journal for serving on an editorial board, and grants to the University of Pennsylvania has from Actelion, United Therapeutics, Gilead, Lung Biotech, and Bayer for CME courses. GMH reports personal fees from George Lehman Group, Genentech, Medna LLC and Patients Like Me. GRW reports support from GlaxoSmithKline, PulmonX and Boerhringer-Ingelheim, GTO reports personal fees from AstraZeneca. RGB reports royalties from UpToDate. DJL has received consulting fees from Genentech/Roche, Boehringer-Ingelheim, Gilead, Pharmakea, Veracyte, Galapagos, Philips Respironics, Patara Pharmaceuticals, Degge Group and the France Foundation related to IPF; Columbia University has received funding for clinical trials in IPF from Boehringer-Ingelheim, Gilead, Bayer, Global Blood Therapeutics and Fibrogen; Columbia University has received funding from the Pulmonary Fibrosis Foundation for DJL’s consulting services.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This research was conducted according to the principles of the Declaration of Helsinki. MESA was reviewed and approved by the Institutional Review Boards (IRB) at each of the participating study sites including Columbia University, Johns Hopkins University, Wake Forest University, Northwestern University, UCLA, the University of Minnesota and was also approved by the IRB at the University of Virginia for genetic analyses. The Framingham Heart Study was approved by the Boston University IRB and the Brigham and Women’s Hospital IRB. The Columbia ILD Study was approved by the Columbia University Medical Center IRB.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
Online Data Supplement for “Genome-wide association study of subclinical interstitial lung disease in MESA”. (DOCX 14799 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Manichaikul, A., Wang, XQ., Sun, L. et al. Genome-wide association study of subclinical interstitial lung disease in MESA. Respir Res 18, 97 (2017). https://doi.org/10.1186/s12931-017-0581-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-017-0581-2