
Abstract
Monoclonal antibodies (mAbs) are a major targeted therapy for malignancies, infectious diseases, autoimmune diseases, transplant rejection and chronic inflammatory diseases due to their antigen specificity and longer half-life than conventional drugs. However, long-term systemic antigen neutralization by mAbs may cause severe adverse events. Improving the selectivity of mAbs to distinguish target antigens at the disease site from normal healthy tissue and reducing severe adverse events caused by the mechanisms-of-action of mAbs is still a pressing need. Development of pro-antibodies (pro-Abs) by installing a protease-cleavable Ab lock is a novel and advanced recombinant Ab-based strategy that efficiently masks the antigen binding ability of mAbs in the normal state and selectively “turns on” the mAb activity when the pro-Ab reaches the proteolytic protease-overexpressed diseased tissue. In this review, we discuss the design and advantages/disadvantages of different Ab lock strategies, focusing particularly on spatial-hindrance-based and affinity peptide-based approaches. We expect that the development of different masking strategies for mAbs will benefit the local reactivity of mAbs at the disease site, increase the therapeutic efficacy and safety of long-term treatment with mAbs in chronic diseases and even permit scientists to develop Ab drugs for formerly undruggable targets and satisfy the unmet medical needs of mAb therapy.
Similar content being viewed by others
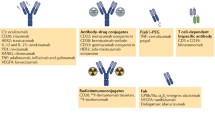


Background
Monoclonal antibodies (mAbs) exist naturally in the human body and have become a mainstream therapeutic option for several kinds of diseases in the clinic, such as autoimmune diseases [24], infectious diseases [22], malignancies [152] and transplant rejection [95]. The pharmaceutical industry has shown continued interest in developing mAb products. This interest is partially driven by the high specificity of mAbs to target antigens as compared with small molecular drugs, their long half-life during systemic circulation, and a well-established, cost-effective platform for producing mAbs to improve the yields, reduce the manufacturing cost and minimize the unexpected safety issues in clinical trials [42, 94, 98, 132, 191]. The first therapeutic mAb, Muromonab-CD3 (also known as Orthoclone OKT3) was a mouse mAb that specifically targets human cluster of differentiation 3 (CD3) on T lymphocytes. It was approved by US Food and Drug Administration (FDA) for treating kidney transplant rejection in 1986. Since then, global sales and the approval rate of mAbs have shown dramatic growth annual (about four products per year). As of December 2019, 79 mAb products had been approved and marketed in the US and Europe and over 300 mAb products were in development [42, 99]. The market for mAbs is expected to continue to grow at a compound annual growth rate (CAGR) of 8% or more annually [42] and worldwide sales of mAb products are estimated to be nearly 300 billion US dollars by 2025 [99]. These figures suggest that mAbs will continue to play a dominant role as a major class of biopharmaceutical products worldwide. However, systemic neutralization of antigens by mAb drug administration carries the risk of a range of adverse events that are associated with the specific target antigens or mechanisms which are essential for the physiological behavior of normal tissues [61, 175].
Systemic administration of mAb drugs may induce severe adverse events by mechanism-of-action-related effects, which means that the mAb drugs eliminate target antigens that maintain physiological functions in normal tissues [61]. For example, in the field of autoimmune disease, mAbs such as Infliximab (Remicade®; Janssen Biotech) or Adalimumab (Humira®; Abbott), work directly against the pro-inflammatory cytokine, tumor necrosis factor α (TNF-α) to treat severe rheumatoid arthritis (RA) [44, 115, 172, 175]. Due to the key role of TNF-α in Mycobacterium tuberculosis (M. tuberculosis) infection and immunity [8, 77], anti-TNF-α therapy increases the frequency of latent tuberculosis reactivation and is also associated with an increased risk of other serious infections and malignancies [41, 140]. It has been reported that progressive multifocal leukoencephalopathy (PML), which is a fatal and rapidly progressive demyelinating disease, has been induced by reactivating latent infection with the polyomavirus John Cunningham virus (JCV) in the central nervous system after treatment with Natalizumab (Tysabri®; Biogen). Natalizumab is a humanized anti-adhesion molecule α4 integrin mAb that is used to treat multiple sclerosis by directly combating T cell trafficking and adhesion [97, 101, 135, 143]. Based on clinical trial data from a 3,147 patient cohort study, the risk frequency of PML corresponds to about 1 to 1000 patients after 18 months of Natalizumab treatment [195] and may originate from immunosuppression of T cell depletion. A similar situation was observed in the field of oncology, when Rituximab (Rituxan®; Genentech), a chimeric anti-CD20 mAb, was used to treat non-Hodgkin’s lymphoma (NHL) by directly eliminating of B cells. The NHL patients receiving Rituximab therapy were reported to have decreased host immunity that triggered PML disease by reactivating JCV [4, 19]. The humanized mAb against CD11a (Efalizumab; Reptiva®; Genentech) was reported to be associated with four PML cases during chronic plaque psoriasis treatment. Suspension of marketing authorization was recommended by the European Medicines Agency (EMA) and phased voluntary withdrawal took place in the United States [20, 114]. Alemtuzumab (Lemtrada®; Campath®; Genzyme), is a humanized anti-CD52 mAb used to avoid immune rejection by bone-marrow [23, 60] and renal [182] transplantation or treat chronic lymphocytic leukaemia (CLL) [91] and multiple sclerosis [67] by depleting CD52-expressing cells, such as CD4+ and CD8+ T cells, monocytes and nature killer cells (NK cells). However, around 3% patients with early multiple sclerosis treated with Alemtuzumab developed serious and fatal thrombocytopaenia [59, 63, 67] and 45% (5 of 11 patients) of patients with peripheral T cell lymphoproliferative disorders showed severe multi-lineage haematopoietic toxicity including lymphopaenia, neutropaenia and thrombocytopaenia [55]. Ipilimumab (Yervoy®; Bristol-Myers Squibb) is a mAb which specifically targets cytotoxic T-lymphocyte-antigen 4 (CTLA-4), one of the major immune checkpoints that regulate adaptive immune responses [47]. It is used to treat metastatic melanoma as a monotherapeutic agent and treat advanced renal cell carcinoma combined with Nivolumab (anti-PD-1 Ab; Opdivo®; Bristol-Myers Squibb) [74]. The CTLA-4 blockage can increase T cell stimulation and activity to continually attack tumor cells [103], but it often causes a range of immune-related adverse events such as rash, hepatitis and even enterocolitis that sometimes requires a colectomy [128, 183]. Humanized anti-HER2/neu mAb (Trastuzumab; Herceptin®; Genentech), used to treat HER2-positive metastatic breast cancer has been reported to possibly also target HER2-expressing cardiomyocytes, block all downstream signaling from HER2 and trigger congestive heart failure risk [170] or cardiac dysfunction in up to 4% patients with Transtuzumab monotherapy with higher incidence in patients receiving additional chemotherapy, such as anthracyclines [78, 129]. In addition, mAbs targeting immune cells (e.g., T or B cells), such as CD3-specific (Muromonab-CD3 )[130], CD20-specific (Rituximab) [188], CD28-specific (TGN1412) [165] or CD52-specific (Alemtuzumab) [186, 187] mAbs, cannot only deplete these targeted cells, but possibly also trigger immediate life-threating cytokine storm (also known as cytokine-released syndrome (CRS)) [29, 79, 135, 165, 185], causing a systemic inflammation response, organ injury and failure even leading to death [165]. As the systemic depletion of antigens may cause unpredictable reactions in patients during Ab therapy, we also list several side effects that have been documented for the above mentioned mAbs (Table 1). Improvement of the selectivity of mAb to distinguish target antigens or cells at the disease site from normal healthy tissue may improve safety and therapeutic efficacy during mAb therapy.
Different masking strategies for pro-antibody drug development
In order to increase the selectivity of mAbs at the disease site to allow them to carry out their function locally, mAb drugs should ignore the target antigen in normal healthy tissue and be preferentially active in the disease region. One way to achieve this goal is by generation of a pro-antibody (pro-Ab) by installing a protease-cleavable Ab lock, which was defined as the molecule that can interfere the antigen binding ability of Ab drugs, is a novel and advanced recombinant Ab-based strategy that selectively “turns on” mAb activity when the pro-Ab reaches proteolytic enzyme (i.e. protease)-overexpressed diseased tissue. A pro-Ab is made up of two essential parts, a “masking domain” that can physically block or interfere with the antigen binding ability of an mAb; and a substrate peptide of disease-associated proteases that connect the masking domain to the N-terminal of the light chain and/or heavy chain of the mAb. The addition of the masking domain results in mAbs with significantly reduced binding ability for their target antigens that, upon exposure to overexpressed proteases at disease site, reactivate the original mAb binding activity, thereby improving the selectivity of the mAb and preventing on-target toxicity during systemic circulation of mAb drugs (Fig. 1). In this review, we will discuss a variety of masking strategies (Table 2), especially spatial hindrance-based (i.e., mask antigen binding ability of mAb by sterically interference) and affinity peptide-based theory (i.e., occupation of an antigen binding site of a mAb by an affinity peptide) in the designing pro-Ab drugs, and discuss their various advantages and disadvantages.

Schematic of pro-antibody selectively activated at the disease region by installing a protease-cleavable Ab lock. (1) Generation of pro-antibody (pro-Ab) by installing a protease-cleavable Ab lock is a novel and advanced recombinant Ab-based strategy that (2) selectively “turns on” the mAb activity when the pro-Ab reaches proteolytic enzyme (i.e., protease)-overexpressed diseased tissue, (3) locally neutralizing the target antigen and reducing on-target toxicity caused by systemic administration of Ab drugs during disease treatment. Ab, antibody; Ag, antigen
Special hindrance-based Ab lock
Autologous hinge domain
Lu and colleagues [100] used an autologous human immunoglobulin G1 (IgG1) hinge as a universal Ab lock to cover the TNF-α-binding site of Infliximab (anti-TNF-α Ab) by linking it with matrix metalloproteinase-2 and -9 (MMP-2/9) substrate (Gly-Pro-Leu-Gly-Val-Arg; GPLGVR) to generate Pro-Infliximab. Once the Pro-Infliximab encounters the overexpressed MMP-2/9 and is specifically hydrolyzed in the disease region of rheumatoid arthritis (RA), the cleaved Pro-Infliximab is specifically activated and neutralizes the target antigen to suppress RA progression (Fig. 2). The Ab lock significantly inhibited the TNF-α-binding ability of Pro-Infliximab by 395-fold as compared with the original Infliximab and MMP-2/9 can completely reactivate the TNF-α neutralizing ability of Pro-Infliximab to block TNF-α-induced nucleus factor kappa B (NF-κB) signaling [100]. Lu et al. also proved that Pro-Infliximab was only selectively and gradually activated at the disease site (i.e., mouse paws) but not other peripheral organs (e.g. peripheral blood, colon, lung or spleen) of a human TNF-α transgenic mouse model, which can spontaneously induce severe chronic arthritis at paws by approximately 20 weeks of age, and presented similar pharmacokinetics (PK) and bio-distribution to Infliximab in healthy DBA/1 mice [100]. Furthermore, Pro-Infliximab not only provided equivalent therapeutic efficacy to Infliximab, but also maintained mouse immunity against Listeria infection in a RA transgenic mouse model, leading to significantly higher survival rate (71%) than that of the Infliximab-treatment group (0%) [100]. In addition, Pro-Infliximab was able to significantly reduce the binding and neutralizing effect of anti-Infliximab idiotypic Ab, which is a major problem in Ab-based therapy after repeat administration of Ab drugs [21], by 108-fold as compared to original Infliximab, and the neutralizing activity of anti-Infliximab idiotypic Ab to Pro-Infliximab could be completely restored after MMP-2/9 cleavage, revealing that the autologous hinge domain forms a spatial barrier and protects antigen binding site of Ab drug from binding of corresponding anti-idiotypic Ab [100]. The spatial-hindrance-based Ab lock has the following advantages: (1) Can be widely applied to a variety of Ab drugs with different target antigens; (2) Autologous hinges may minimize the risk of immunogenicity of anti-Hinge Ab from the host; (3) Ab lock can significantly inhibit the binding and neutralizing ability of anti-idiotypic Ab to original Ab drugs, at least in the case of Infliximab; (4) The changeable design of protease substrate between Ab lock and Infliximab can be applied to any alternative Ab drugs that are used for RA or even other disease treatments. It is expected that a protease cleavable and efficient Ab lock can significantly increase the selective activation of Ab drugs at the disease site and reduce the on-target toxicities of Ab drugs during systemic circulation, thus potentially improving the clinical benefit and quality of life of patients.

The autologous hinge domain as a universal Ab lock for enhancing disease site selectivity and minimizing the on-target toxicity of Ab drugs. Lu et al. used an autologous human IgG1 hinge domain as an Ab lock to cover the antigen binding site of a Ab drug (i.e., anti-TNF-α Ab, Infliximab) by using the MMP-2/9 substrate linker to generate a pro-Ab. When the pro-Ab encounters activated protease at the inflamed region (e.g., the RA region), the substrate linker is hydrolyzed, the Ab lock is released, and the pro-Ab can specifically activate and neutralize the target antigen (e.g., TNF-α) at the disease site to inhibit RA progression. Ab, antibody; IgG1, immunoglobulin G1; MMP, matrix metalloproteinase; TNF-α, tumor necrosis factor α
Lu et al. used an autologous human IgG1 hinge domain as an Ab lock to cover the antigen binding site of a Ab drug (i.e., anti-TNF-α Ab, Infliximab) by using the MMP-2/9 substrate linker to generate a pro-Ab. When the pro-Ab encounters activated protease at the inflamed region (e.g., the RA region), the substrate linker is hydrolyzed, the Ab lock is released, and the pro-Ab can specifically activate and neutralize the target antigen (e.g., TNF-α) at the disease site to inhibit RA progression. Ab, antibody; IgG1, immunoglobulin G1; MMP, matrix metalloproteinase; TNF-α, tumor necrosis factor α.
Coiled-coil domain
Trang and colleagues [176] also developed a generalizable approach for Ab masking with a leucine-rich and parallel heterodimeric coiled-coil domain that spatially occupies the antigen binding site of Ab drugs (Fig. 3). The coiled-coil (CC) masking domains with different inter-coil affinities or orientations were derived from de novo designs [173] or native human proteins [15, 149], such as c-Fos and c-Jun. In the initial test of this strategy, the authors evaluated the masking efficiency of parallel heterodimeric coils with low (CC1) and high (CC2A and 2B) inter-coil interaction [173], disulfide-linked coils (CC3) [149], antiparallel heterodimeric (CC4) [106] and helix-turn-helix homodimeric coils (CC5) [131] to a CD19-binding Ab hBU12 [53] by assessing the antigen binding ability to CD19-expressing Raji cells. The results suggested that parallel heterodimeric coiled-coils CC2A, CC2B and CC3 had excellent blocking activity of over 300-fold. In particular, CC2B was found more suitable for further developing Ab-drug conjugates (ADCs) because the lack of covalent disulfide linkages between the coiled-coil domains minimizes the heterogeneity of chemical drug attachment by using endogenous disulfide conjugation. Trang further linked CC2B peptide to all four N-terminals (two light chains and two heavy chains) of different Ab drugs, including Rituximab (anti-CD20 mAb) [80], Trastuzumab (anti-HER2 mAb) [105], h15H3 (anti-αVβ6 mAb) [176] and 145-2C11 (anti-mouse CD3 mAb) [88] with MMP-2 and MMP-9 cleavable sequence (PLGLAG [72] or IPVSLRSG [178]) and proved that the CC2B domain can significantly decrease the antigen binding affinity of each mAb at least 80-fold, 470-fold, 290-fold and 1000-fold, respectively. After treatment with purified MMP-2, the CC2B mask can be efficiently removed to restore the antigen binding ability of the Ab drugs to within 1.7-fold as compared to parent Ab. With ADCs, the incorporation of a protease-cleavable sequence between the coiled-coil mask and Ab drugs significantly improved the selectivity of Ab drugs to the disease site from normal healthy tissues, thereby increasing the pharmacokinetics and therapeutic efficacy by avoiding the effect of antigen sink in a xenograft tumor mouse model. Furthermore, the sterical-hindrance-based coiled-coil mask also has the following advantages: (1) Possible wide application to different Ab drugs in the clinic or in development; (2) Coiled-coil mask provides high masking efficiency to Ab drugs in a condition that lacks proteolytic enzymes and maintains the Ab level by avoiding the effect of antigen sink; (3) High release rate of the coiled-coil mask from Ab drugs after cleaving with disease-specific protease. However, the relatively complex helix structure and custom mutated coiled-coil mask may significantly reduce the productive rate and increase the risk of immunogenicity during long-term systemic administration of coiled-coil masked-Ab drugs.

The coiled-coil domain as a universal Ab lock for enhancing the selectivity of Ab drugs to the disease site. Trang et al. used leucine-rich and parallel heterodimeric coiled-coil domains as an Ab lock that spatially occupies the antigen binding site of Ab drugs (i.e., anti-CD19 mAb, anti-CD20 mAb, anti-HER2 mAb, anti-αVβ6 mAb and anti-mouse CD3 mAb) by using MMP-2 or -9 substrate linker (PLGLAG or IPVSLRSG) to generate a pro-Ab. When protease activation occurs at the inflamed region (e.g., tumor region), the coiled-coil domain is released, and the pro-Ab can specifically revive and neutralize the target antigen (e.g., CD19, CD20, HER2, αVβ6 or CD3) at the disease site to inhibit disease progression. Ab, antibody; CD, cluster of differentiation; HER2, human epidermal growth factor receptor 2; MMP, matrix metalloproteinase
Trang et al. used leucine-rich and parallel heterodimeric coiled-coil domains as an Ab lock that spatially occupies the antigen binding site of Ab drugs (i.e., anti-CD19 mAb, anti-CD20 mAb, anti-HER2 mAb, anti-αVβ6 mAb and anti-mouse CD3 mAb) by using MMP-2 or − 9 substrate linker (PLGLAG or IPVSLRSG) to generate a pro-Ab. When protease activation occurs at the inflamed region (e.g., tumor region), the coiled-coil domain is released, and the pro-Ab can specifically revive and neutralize the target antigen (e.g., CD19, CD20, HER2, αVβ6 or CD3) at the disease site to inhibit disease progression. Ab, antibody; CD, cluster of differentiation; HER2, human epidermal growth factor receptor 2; MMP, matrix metalloproteinase.
Non-antibody protein fragment
Chen and colleagues [27] developed protease-activated pro-Ab by masking the antigen binding site of anti-EGFR Ab (Cetuximab) or anti-TNF-α Ab (Infliximab), respectively, with latency-associated peptide (LAP) derived from transforming growth factor-β (TGF-β) and connected it to the N-terminal of heavy chain of Ab by a substrate peptide for MMP-2 (i.e., GPLGVR) (Fig. 4). The LAP domain reduced the antigen binding activity of anti-EGFR Ab and anti-TNF-α Ab by 53.8 and 53.9%, respectively. After MMP-2 digestion of LAP-masked anti-EGFR Ab or LAP-masked anti-TNF-α Ab, the antigen binding ability rose progressively to levels similar to that of the unmodified Ab. The advantages of this masking strategy are that (1) the LAP domain derived from endogenous TGF-β proteins may reduce the opportunity of immunogenicity from the host; (2) the inhibitory domain displays no apparent or known biological function other than blocking the activity of the original proteins and can minimize unpredictable adverse events or cross reaction to other proteins. However, the poor masking efficiency of the LAP domain (only masks 1.86-fold) to the EGFR, or TNF-α binding ability of the original Ab drugs, and the reduced productive yield (retained only 33% productive yield as compared with original Ab) affected by significantly increased molecular weight (MW) of LAP-masked Ab (approximately 240 kDa) may increase difficulty in obtaining enough pro-Abs to investigate the PK, bio-distribution, therapeutic efficacy and on-target toxicities in living disease models and limit its broad application to other Ab drugs.

Endogenous LAP peptide is a widely applied Ab lock for enhancing the selectivity of Ab drugs to the disease site. Chen et al. used latency-associated peptide (LAP) derived from transforming growth factor-β (TGF-β) as a widely applied Ab lock and connected it to the N-terminal of the heavy chain of Ab drugs (e.g., the anti-EGFR Ab, Cetuximab, or the anti-TNF-α Ab, Infliximab) by a substrate peptide for MMP-2 (i.e., GPLGVR). The LAP domain reduced the antigen binding activity of Ab drugs and was selectively activated by MMP-2 for specific antigen targeting at the disease site. Ab, antibody; EGFR, epidermal growth factor receptor; TNF-α, tumor necrosis factor α; MMP, matrix metalloproteinase
Chen et al. used latency-associated peptide (LAP) derived from transforming growth factor-β (TGF-β) as a widely applied Ab lock and connected it to the N-terminal of the heavy chain of Ab drugs (e.g., the anti-EGFR Ab, Cetuximab, or the anti-TNF-α Ab, Infliximab) by a substrate peptide for MMP-2 (i.e., GPLGVR). The LAP domain reduced the antigen binding activity of Ab drugs and was selectively activated by MMP-2 for specific antigen targeting at the disease site. Ab, antibody; EGFR, epidermal growth factor receptor; TNF-α, tumor necrosis factor α; MMP, matrix metalloproteinase.
Ab fragment
Metz et al. [108] engineered a trivalent bi-specific Ab by linking a disulfide-stabilized variable fragment (dsFv), which is specific against c-Met antigen that is deregulated in several types of cancer [110], to the C-terminus of the heavy chain of a bivalent anti-HER3 Ab with one-armed protease substrate peptide (GPLGMLSQ, GPLGLWAQ and GPLGIAGQ for MMP-2 and MMP-9 [118], and GGGRR for urokinase plasminogen activator (uPA) [28]). In this design, the c-Met binding ability of dsFv was sterically interfered with by the Fc portion of the front anti-HER3 Ab. After proteolytic processing by the corresponding protease, the dsFv is swiveled open and recovers its c-Met binding activity (Fig. 5a). The authors were able to illustrate an approximate 1000-fold difference in affinity between the pro-Ab format and the protease-activated form in vitro, suggesting that this trivalent bi-specific Ab may enhance tumor targeting as a result of hydrolyzation by proteases overexpressed in tumor microenvironment.

Ab fragment as an Ab lock for shielding antigen binding ability of specific Ab drugs. a Metz et al. constructed a trivalent bi-specific Ab by linking a disulfide-stabilized variable fragment (dsFv), which is specific against one antigen (e.g., c-Met), to the C-terminus of the heavy chain of a whole Ab (e.g., anti-HER3 Ab) with a one-armed protease substrate peptide (e.g., GPLGMLSQ, GPLGLWAQ and GPLGIAGQ for MMP-2 and MMP-9 or GGGRR for urokinase plasminogen activator (uPA)). In this design, the antigen binding ability of dsFv was sterically interfered with by the Fc portion of the front whole Ab until protease cleavage and reactivation of the dsFv activity. b Onuoha’s and Pai’s groups developed an activatable dual variable domain (aDVD) Ab by linking the dsFv from one Ab (e.g., anti-ICAM-1 Ab or anti-PSCA Ab) to the N-terminus of light and heavy chains of another Ab (e.g., anti-TNF-α Ab or anti-CTLA-4 Ab) with MMP-1- (PLGLWA) or MT-SP1-cleavable sequence (LSGRSDNH). The outer dsFv can significantly shield the antigen binding ability of the inner Ab and lead the aDVD to the disease site (e.g., tumor site), to be reactivated by disease-specific proteases and restore the antigen neutralizing ability of inner Ab drugs. Ab, antibody; HER3, human epidermal growth factor receptor 3; TNF-α, tumor necrosis factor α; MMP, matrix metalloproteinase; MT-SP1, activated membrane type-serine protease 1; ICAM-1, intercellular adhesion molecule 1; CTLA-4, cytotoxic T-lymphocyte-antigen 4
Similar to the strategy of activatable trivalent bi-specific Ab, Onuoha and colleagues [124] developed an activatable dual variable domain (aDVD) Ab by linking the dsFv from Ab against intercellular adhesion molecule 1 (ICAM-1) to the N-terminus of light and heavy chain of anti-TNF-α Ab (Infliximab and Adalimumab) with MMP-1-cleavable sequence (PLGLWA) (Fig. 5b). In this strategy, the outer arm of the anti-ICAM-1 variable domain retained its antigen binding ability and significantly masked the TNF-α binding ability of Infliximab or Adalimumab in the inner arm by greater than 1000-fold in KD as measured by surface plasmon resonance (SPR) in vitro. The authors further proved that the human synovial fluid and physiologic concentrations of MMP-1 enzyme sufficiently cleaved and restored the TNF-α binding ability of aDVD and locally accumulated aDVD in the disease region. Pai et al. [125] also applied the aDVD strategy to anti-CTLA-4 Ab, which is an immune checkpoint inhibitor that specifically neutralizes CTLA-4 on activated T cells, enhancing the anti-cancer immunity, and forming anti-CTLA-4 aDVD. The CTLA-4 binding domain of the inner anti-CTLA-4 Ab is shielded by an outer dsFv specifically targeting the prostate stem cell antigen (PSCA) and linking with an activated membrane type-serine protease 1 (MT-SP1)-cleavable sequence (LSGRSDNH) (Fig. 5b). The outer anti-PSCA dsFv can significantly shield the CTLA-4 binding ability of the inner Ab and selectively increase CTLA-4-neutralizing activity by 25-fold as compared to the intact aDVD form after MT-SP-1 cleavage. The anti-CTLA-4 aDVD exhibits a potent anti-cancer effect by increasing tumor-infiltrating CD8+ T cells and decreasing the tumor-infiltrating Treg population in allogenic tumor-bearing mice and further preventing treatment-induced multiorgan toxicity. As with the activatable bi-specific Ab, the substrate linker of aDVD can also be specifically hydrolyzed by overexpressed protease in the inflamed region, restore the antigen neutralizing ability (e.g., TNF-α or CTLA-4) and potentially enhance the therapeutic index of Ab drugs for the treatment of RA, malignancies and other inflammatory diseases in the future. However, the antigen binding ability of the outer domain of trivalent bi-specific Ab or aDVD may still contribute to undesirable targeting of antigens by the masked Ab to non-diseased tissue and further induce unpredictable adverse events.
(A) Metz et al. constructed a trivalent bi-specific Ab by linking a disulfide-stabilized variable fragment (dsFv), which is specific against one antigen (e.g., c-Met), to the C-terminus of the heavy chain of a whole Ab (e.g., anti-HER3 Ab) with a one-armed protease substrate peptide (e.g., GPLGMLSQ, GPLGLWAQ and GPLGIAGQ for MMP-2 and MMP-9 or GGGRR for urokinase plasminogen activator (uPA)). In this design, the antigen binding ability of dsFv was sterically interfered with by the Fc portion of the front whole Ab until protease cleavage and reactivation of the dsFv activity. (B) Onuoha’s and Pai’s groups developed an activatable dual variable domain (aDVD) Ab by linking the dsFv from one Ab (e.g., anti-ICAM-1 Ab or anti-PSCA Ab) to the N-terminus of light and heavy chains of another Ab (e.g., anti-TNF-α Ab or anti-CTLA-4 Ab) with MMP-1- (PLGLWA) or MT-SP1-cleavable sequence (LSGRSDNH). The outer dsFv can significantly shield the antigen binding ability of the inner Ab and lead the aDVD to the disease site (e.g., tumor site), to be reactivated by disease-specific proteases and restore the antigen neutralizing ability of inner Ab drugs. Ab, antibody; HER3, human epidermal growth factor receptor 3; TNF-α, tumor necrosis factor α; MMP, matrix metalloproteinase; MT-SP1, activated membrane type-serine protease 1; ICAM-1, intercellular adhesion molecule 1; CTLA-4, cytotoxic T-lymphocyte-antigen 4.
Affinity-peptide based Ab lock
Affinity peptide
The most rapidly progressing affinity-based pro-Ab strategy is PROBODY therapeutics developed by CytomX therapeutics, Inc. The target binding region of the mAb is masked by an affinity peptide screened out using phage display library technology and linked through a protease substrate peptide which allows it be cleaved in disease microenvironment (Fig. 6). The Probody therapeutic platform ensures the Ab drugs can only be activated in diseased tissue but not in normal healthy tissues, thereby preventing unpredictable side effects caused by systemic reactions in traditional Ab drugs. For example, Desnoyers and colleagues generated an epithelial growth factor receptor (EGFR) pro-Ab by selecting a binding peptide from a bacterial peptide display library that could specifically mask the antigen-binding site of anti-EGFR Ab (Cetuximab, Erbitux®) and linking with substrate peptide of tumor-associated protease, urokinase-type plasminogen activator (uPA) [35]. The EGFR pro-Ab exhibits 48-fold weaker antigen binding ability than the original Cetuximab, maintains an intact format in systemic circulation and the biological activity could be specifically restored in the tumor microenvironment in a protease dependent manner. The authors also proved that the EGFR Probody therapeutic can increase drug exposure in xenograft tumors by avoiding target-mediated drug disposition (TMDD) in mouse models, reduce the skin toxicity associated with parental Cetuximab and elevate the safety index 3- to 15-fold during Probody treatment in cynomolgus monkeys. Similar strategies were also applied to other Abs such as anti-vascular cell adhesion molecule 1 (VCAM-1) mAb [43] and anti-EGFR mAb (Panitumumab, Vectibix®) [193]. Furthermore, Liu et al. [93] and Yang et al. [192] applied the Probody approach to the maytansine (DM1)-conjugated anti-EGFR mAb (Panitumumab) and generated a pro-antibody-drug conjugate (PDC) named PanP-DM1. Regardless of DM1 conjugation, the PanP-DM1 still displayed 12-fold weaker binding ability to immobilized EGFR antigen than parental Panitumumab and significantly improved the cancer-selective activity, therapeutic and safety index over traditional ADC. Collectively, the mouse and cynomolgus monkey data indicate that the Probody therapeutics can significantly expand the therapeutic window of mAb therapy. However, these exogenous and affinity-based masking peptides may not be efficiently released from pro-antibodies after protease cleavage due to the binding ability of masking peptides to antibodies, and the exogenous property may also increase the risk of undesirable immune responses from the host. In addition, the customized masking peptide may limit the Probody approach from being widely applied to different Ab drugs in the development stage or in the clinic.

Affinity peptide-based Ab lock for masking the antigen binding activity of Ab drugs. CytomX therapeutics, Inc. developed PROBODY therapeutics by masking the antigen binding site of Ab drugs (e.g., anti-EGFR Ab, anti-VCAM-1 Ab or anti-EGFR ADC) with an affinity peptide screened out from phage display library technology and linked it to an Ab through a protease substrate peptide (e.g., LSGRSDNH for uPA and MT-SP1). The Pro-Ab exhibits weaker antigen binding ability than the original Ab drug, maintains intact format in systemic circulation and the biological activity could be specifically restored in the tumor microenvironment in a protease dependent manner. Ab, antibody; EGFR, epidermal growth factor receptor; ADC, antibody-drug conjugate; VCAM-1, vascular cell adhesion molecule 1; uPA, urokinase plasminogen activator; MT-SP1, activated membrane type-serine protease 1
CytomX therapeutics, Inc. developed PROBODY therapeutics by masking the antigen binding site of Ab drugs (e.g., anti-EGFR Ab, anti-VCAM-1 Ab or anti-EGFR ADC) with an affinity peptide screened out from phage display library technology and linked it to an Ab through a protease substrate peptide (e.g., LSGRSDNH for uPA and MT-SP1). The Pro-Ab exhibits weaker antigen binding ability than the original Ab drug, maintains intact format in systemic circulation and the biological activity could be specifically restored in the tumor microenvironment in a protease dependent manner. Ab, antibody; EGFR, epidermal growth factor receptor; ADC, antibody-drug conjugate; VCAM-1, vascular cell adhesion molecule 1; uPA, urokinase plasminogen activator; MT-SP1, activated membrane type-serine protease 1.
Cross-masking antibodies
The cross-masking Ab approach involves mutual masking of two different Ab drugs. In this design, one Ab drug is linked with a corresponding antigen epitope of another Ab drug via protease-specific substrate peptide and vice versa (Fig. 7). Donaldson and colleagues [36] conducted a proof-of-concept study for this strategy in vitro by using two single chain Fv (scFv) derived from two anti-EGFR Abs (Cetuximab and Matuzumab) and linking with a point mutated soluble EGFR domain III (sEGFRdIII) (S460P/G461N for masking Cetuximab and Q384A/Q408M/H409E for masking Matuzumab) through MMP-9 substrate peptide, respectively. The mutation of sEGFRdIII significantly reduced the binding affinity of linked anti-EGFR scFv from 420 ± 270 nM to 3500 ± 1.1 nM and increased the releasing efficiency from cross-masking Ab after MMP-9 cleavage. After purifying and mixing together individual constructs, it allowed the assembly of the cross-masking Ab complex. The SPR and Flow cytometry data suggested that the cross-masking Abs poorly interacted (approximately 8.3-fold weaker than parental scFv) with native EGFR antigen and the binding ability was restored after MMP-9 treatment as compared with the original anti-EGFR scFv. The cross-masking strategy permits the simultaneous delivery of two Ab drugs that synergize or independently target two different tumor-associated antigens and may increase the therapeutic efficiency of diseases. However, the low masking efficiency, structural complexity, and homogeneity of cross-masking Ab formation and customized mutation and unpredictable releasing rate of the masking domain may limit its wide application to different Ab drugs.

Cross-masking antibodies for limiting off-target effects of Ab drugs. Donaldson et al. used a cross-masking Ab approach to simultaneously block two Ab drugs. In this design, one Ab drug was linked with a corresponding antigen epitope (e.g., mutated soluble EGFR domain III (sEGFRdIII)) of another Ab drug via a protease-specific substrate peptide (VPLSLYS for MMP-9) and vice versa. The mutated masking epitope can significantly reduce the binding affinity of the linked Ab drug to a specific antigen and increase the releasing efficiency from the cross-masking Ab after MMP-9 cleavage, thereby restoring the antigen-neutralizing activity of the Ab drug. Ab, antibody; EGFR, epidermal growth factor receptor; MMP, matrix metalloproteinase
Donaldson et al. used a cross-masking Ab approach to simultaneously block two Ab drugs. In this design, one Ab drug was linked with a corresponding antigen epitope (e.g., mutated soluble EGFR domain III (sEGFRdIII)) of another Ab drug via a protease-specific substrate peptide (VPLSLYS for MMP-9) and vice versa. The mutated masking epitope can significantly reduce the binding affinity of the linked Ab drug to a specific antigen and increase the releasing efficiency from the cross-masking Ab after MMP-9 cleavage, thereby restoring the antigen-neutralizing activity of the Ab drug. Ab, antibody; EGFR, epidermal growth factor receptor; MMP, matrix metalloproteinase.
Bivalent peptide-double strand DNA (dsDNA) conjugates
Janssen and colleagues generated a protease-activatable bivalent peptide-double strand DNA (dsDNA) conjugate as an Ab masking molecule to achieve a reversible blocking effect on antigen binding ability of specific Ab drugs [70]. In this design, the masking molecule contains two binding epitopes (ELDRWEKIRLRP) of anti-HIV p17 mAb and conjugates to a 35 base-paired single strand DNA (ssDNA) by linking with a maleimide functionalized MMP-2-cleavable sequence (maleimide-PLGLAG). The two complementary ssDNA fragments can naturally hybrid to form a double helix and effectively bridge the masking epitope between two antigen-binding sites within the same Ab, forming an Ab-ligand complex (Fig. 8). Janssen proved that the bivalent peptide-dsDNA conjugates can efficiently mask the p17 binding affinity by 8-fold of the model anti-HIV p17 mAb as compared to parental Ab, and the blockage effect of bivalent peptide-dsDNA conjugates was nearly completely restored after cleaving with MMP-2 within 2 h in an in vitro binding assay. However, the low masking efficiency, heterogeneity of Ab-ligand complex formation after mixing constant ratio of bivalent masking molecule and Ab drugs, customized masking epitope, immunogenicity and uncertainty releasing rate of masking peptide may restrict its broad application to various Ab drugs.

Bivalent peptide-double strand DNA (dsDNA) conjugates as affinity-based Ab lock for blocking antigen binding ability of Ab drugs. Janssen et al. generated a protease-activatable bivalent peptide-double strand DNA (dsDNA) conjugate as a masking molecule of Ab for reversible blocking effect on antigen binding ability of specific Ab drugs. In this design, the masking molecule contains two binding epitopes of a specific Ab drug (e.g., anti-HIV p17 mAb) and conjugates to a 35 base-paired single strand DNA (ssDNA) by linking with a maleimide functionalized MMP-2-cleavable sequence (e.g., maleimide-PLGLAG). The two complementary ssDNA fragments can naturally hybridize to form double helix and effectively bridge the masking epitope between two antigen binding sites within the same Ab, forming an Ab-ligand complex. After MMP-2 cleavage in the disease region, the masking molecule is released and the Pro-Ab is also reactivated and performs its original therapeutic function. mAb, monoclonal antibody; HIV, human immunodeficiency virus; MMP, matrix metalloprotease
Janssen et al. generated a protease-activatable bivalent peptide-double strand DNA (dsDNA) conjugate as a masking molecule of Ab for reversible blocking effect on antigen binding ability of specific Ab drugs. In this design, the masking molecule contains two binding epitopes of a specific Ab drug (e.g., anti-HIV p17 mAb) and conjugates to a 35 base-paired single strand DNA (ssDNA) by linking with a maleimide functionalized MMP-2-cleavable sequence (e.g., maleimide-PLGLAG). The two complementary ssDNA fragments can naturally hybridize to form double helix and effectively bridge the masking epitope between two antigen binding sites within the same Ab, forming an Ab-ligand complex. After MMP-2 cleavage in the disease region, the masking molecule is released and the Pro-Ab is also reactivated and performs its original therapeutic function. mAb, monoclonal antibody; HIV, human immunodeficiency virus; MMP, matrix metalloprotease.
Comparison of different masking strategies of Pro-Abs
Development of approaches to address on-target toxicity caused by systemic neutralization of antigens by Ab drugs has generated a new vision in Ab drug development. The generation of Pro-Abs by installing a protease-cleavable Ab lock is a novel and advanced recombinant Ab-based strategy for selective activation of Ab drugs at the disease site. For good design of an Ab lock, multiple factors should be considered, including the masking efficiency, immunogenicity, wide applicability, Ab lock release efficiency after protease cleavage and reduction of side effects (Table 3). Spatial-hindrance-based Ab locks, such as autologous hinge domain, coiled-coil domain, LAP domain or trivalent/tetravalent Ab-based approaches, provide high masking efficiency, high release rate of masking domain and high restoration of antigen binding activity of mAbs after protease cleavage and nearly universal application of various Ab drugs. Affinity-based masking strategies (i.e., masking dependent on affinity peptides or mutant antigens) demonstrate acceptable masking efficiency; however, custom design of masking peptides used in individual mAbs and uncertain release rate of the masking molecule after protease hydrolysis may make it impractical for clinical applications. Depending on the source of the masking domain of Pro-Abs, the Ab lock is involved in the intracellular (i.e., c-Fos and c-Jun) or artificial coiled-coil domain, exogenous Ab fragment in trivalent/tetravalent Ab-based approaches, masking peptide derived from phage display and mutated antigen may increase the risk of immunogenicity during Pro-Ab treatment. In principle, both masking theories (i.e., spatial-hindrance-based or affinity peptide-based approaches) can inhibit the neutralizing and cleaning effect of anti-idiotypic anti-drug Abs, which is one of major problems in Ab therapy, caused by repeated administration of mAb. Lu’s team provided evidence that an autologous hinge can efficiently prevent pro-Infliximab from the neutralizing activity of anti-idiotypic Ab and the TNF-α binding ability can be completely restored by MMP-2/9 cleavage. Most importantly, research groups that developed autologous hinge domain-based, Ab fragment in trivalent/tetravalent Ab-based and phage display-derived masking peptide-based approaches provide evidence that these three Ab lock can efficiently mask antigen binding ability of Ab drugs, and prevent systemic antigen targeting, thereby significantly reducing the side effects caused by the mechanism-of-action of Ab drugs. In summary, there are different advantages with different masking strategies of Pro-Ab; they may provide scientists more options for developing next generation Ab drugs corresponding to the conditions of different type of diseases.
Disease-specific proteases locally unlock and reactivate pro-antibodies in the disease region
Proteases are an attractive target for drug development and deregulation of protease activity has been involved in the pathogenesis of a variety of diseases including malignancies, autoimmune diseases and inflammatory disorders (Table 4). In the field of oncology, for example, the expression of MMP-2 and MMP-9, which are members of the endopeptidase family that can degrade different components of the extracellular matrix (ECM) [87], are upregulated by acute lymphoblastic leukemia (ALL) cells and hydrolyze the basement membrane of blood vessels, releasing matrix-bound vascular endothelial growth factor (VEGF), inducing angiogenesis and stimulating extramedullary infiltration by lymphoblasts [133]. Similarly, MMP-9 plays an important role in migration and survival, and speeds up the disease progression of chronic lymphocytic leukemias (CLL) [11]. MMP-2, MMP-9 and MMP-13 are also reported to be overexpressed in breast cancer patients and correlated with more aggressive phenotypes of breast cancer, poor prognosis and decreased life expectancy [49, 138, 141]. Cathepsin B has been reported to be highly expressed in breast cancer patients and mainly responsible for degrading collagen, fibronectin, laminin and proteoglycans, thereby promoting nodal metastasis [48, 120, 158]. The urokinase plasminogen activator (uPA) system, which represents a family of serine protease, also participates to tumorigenesis. uPA has been reported to be overexpressed in several type of cancers, such as esophageal [156], gastric [71], colorectal [159], pancreatic [119], breast [40, 96, 162], cervical [82], ovarian [90], prostate [134], leukemia [148], brain [64] and renal cancers [168]. It can degrade the basement membrane or ECM around the tissue by itself or further activate the downstream MMP family [33]. The higher uPA level in cancer patients increases the frequency of metastasis and leads to poor prognosis and survival rate [33]. MMPs also serve as inflammatory markers that are involved in the pathogenesis of several autoimmune disorders or chronic conditions. For instance, the expression level of MMP-2 and MMP-9 are increased in the synovial liquid of patients with RA and contribute to the destruction of the cartilage, tendons, and bone in the synovial joints [9, 16, 164, 194]. MMP-1, MMP-8, MMP-9 and MMP-12 have been shown to be upregulated in the disease region of chronic obstructed pulmonary disease (COPD), which is a chronic inflammatory airway disorder commonly caused by cigarette smoking and occupational inhalation (e.g., dust or chemicals) [139]. The overexpressed MMP may increase the ECM degradation in the lungs and therefore accelerate the loss of lung function [142, 155]. It is notable that deregulation of protease activity is an important hallmark of most diseases because it may be required for tissue repair or disease progression [1, 3, 5, 7, 10, 12, 14, 17, 25, 26, 31, 32, 38, 39, 45, 46, 50, 52, 56,57,58, 62, 68, 75, 76, 83, 84, 86, 89, 92, 104, 107, 109, 112, 117, 121,122,123, 126, 127, 136, 144, 150, 151, 153, 157, 160, 163, 166, 167, 174, 177, 184, 189, 190, 196,197,198]. Based on the changeable design of the pro-Ab therapeutic strategy, it can be widely applied to different kinds of diseases by replacing the substrate linker on pro-Ab to correspond to the substrate sequence of specific proteases, which are overexpressed in the disease of interest.
The pro-antibody strategy avoids the inhibitory effect of anti-idiotypic anti-drug antibodies
A major limitation of mAb therapy is generation of anti-idiotypic Abs after repeated administration of Ab drugs. Despite the development of humanized and fully human mAbs that try to minimize immunogenic activity, human anti-human idiotypic Abs (also known as anti-idiotypic Abs) are still generated against the idiotypic domain on the antigen binding site of mAbs. For example, about 12% of RA patients received Adalimumab (a fully human IgG1 mAb against human TNF-α) treatment that tested positive for ADA against Adalimumab [113, 179]. This kind of ADA may result in the formation of immune complexes, which accelerate its clearance from circulation, thereby reducing the drug’s half-life or directly blocking the binding ability of Adalimumab to its target, reducing treatment efficacy. Van Schouwenburg and colleagues even suggest that at least 98% of anti-Adalimumab Abs are capable of neutralizing this agent [180]. Davda et al. indicated that the low incidence of ADA (0–12.7%), which contains 0–0.8% neutralizing ADA, was found following a single-agent treatment with immune checkpoint inhibitors such as anti-PD-1 mAbs (Nivolumab, Pembrolizumab, and Cemiplimab); the anti-CTLA-4 mAb (Ipilimumab); and the anti-PD-L1 mAbs (Avelumab and Durvalumab) in patients with advanced malignancies [2, 34]. However, a high incidence of ADA (23.8–37.8%), which contains 0.5–4.6% neutralizing ADA, was observed against Nivolumab when combining an anti-PD-1/PD-L1 and an anti-CTLA-4 mAb in patients with advanced solid tumors [34]. Fortunately, there were no clinically relevant effects on the safety, pharmacokinetics, or therapeutic efficacy of Nivolumab in patients who developed anti-Nivolumab Abs [34]. It should be noted that the immunogenicity of mAbs is not a simple issue about the sequence homology to human Abs. In the study from Lu et al., the spatial-hindrance-based Ab lock (autologous Hinge domain) significantly reduces the anti-Infliximab idiotypic (anti-I-Id) Ab binding to Pro-Infliximab by 108-fold as compared to Infliximab, and the TNF-α neutralizing ability of Pro-Infliximab can be completely restored after MMP-2/9 cleavage, thereby blocking TNF-α downstream signaling [100]. These data indicate that the pro-Ab strategy may prevent the neutralizing and cleaning effect of anti-idiotypic Abs on Ab drugs, prolong the half-life of Ab drugs and in the future may potentially become an alternative therapeutic option in patients, who had already developed Ab drug-specific anti-idiotypic Abs.
Improving the masking efficacy of pro-antibody by computer modeling
The masking efficiency of different Ab locks, especially spatial-hindrance-based Ab locks, depends on the occupied site or interference range of the masking domain in front of antigen binding site of Ab drugs. The autologous hinge developed by Lu et al. has been proved to widely mask anti-TNF-α Ab (Infliximab), anti-IL-6 receptor Ab, anti-IL-1β Ab, anti-PD-1 Ab and anti-CTLA-4 Ab by 395-fold, 50.3-fold [100], 68-fold, 256-fold and 106-fold (data not shown), respectively. The coiled-coil domain suggested by Seattle Genetics. Inc. can broadly and efficiently block the antigen binding ability of anti-CD20 mAb (Rituximab), anti-HER2 mAb (Trastuzumab), anti-αVβ6 mAb (h15H3) and anti-mouse CD3 mAb (145-2C11) at least 80-fold, 470-fold, 290-fold and 1000-fold, respectively [176]. The LAP domain screened out by Chen et al. can also inhibit the antigen binding activity of anti-EGFR Ab (Cetuximab) or anti-TNF-α Ab (Infliximab) both by 1.86-fold [27]. However, even masking with the same Ab lock, there is inconsistent masking efficiency between different target mAbs. We reasonably think that there is still slight structural difference at the complementary-determining regions (CDRs) or N-terminal end of different mAbs despite the high structural conservation of Abs between different species, leading to the inconsistent masking effect of Ab locks on the binding activity of Ab drugs. In order to optimize the masking efficiency of the Ab lock to different mAbs, adjusting the linker sequence between the Ab lock and Ab drugs by amino acid addition, deletion or substitution, and combining with structure-based computational simulation (such as structure building by using Discovery Studio (San Diego, CA, USA) or molecular dynamic simulation by using Amber software) may assist scientists in rationally predicting and adjusting the length or bending angle of the connecting linker and optimizing the coverage rate of different Ab locks to obtain the highest masking efficiency for various Ab drugs.
Conclusions
Monoclonal Abs continue to be the most rapidly growing and valuable drug field in the pharmaceutical industry due to their high specificity and affinity to target antigens and longer half-life than conventional drugs. However, there is still an urgent need to improve the selectivity of mAbs to distinguish target antigens at the disease site from normal healthy tissue, thereby reducing the severe adverse events caused by mechanism-of-action-related effects of mAbs and elevating its safety and therapeutic efficacy during systemic administration. The development of masking approaches to Ab drugs can improve the local reactivity of mAbs at the disease site, increase the therapeutic efficacy and safety of long-term treatment with mAbs in chronic diseases, and even serve as an alternative therapeutic option for preventing the functional interference of anti-idiotypic Abs. Masking approaches can also permit scientists to develop next generation Ab drugs for targets that have been hitherto undruggable and satisfy the unmet medical needs of mAb therapy.
Availability of data and materials
Not applicable.
Abbreviations
- TNF-α:
-
Tumor necrosis factor α
- CD:
-
Cluster of differentiation
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- HER2:
-
Human epidermal growth factor receptor 2
- HER3:
-
Human epidermal growth factor receptor 3
- NK:
-
Natural killer cell
- Treg:
-
Regulatory T cell
- PML:
-
Progressive multifocal leukoencephalopathy
- CRS:
-
Cytokine-released syndrome
- Ab:
-
Antibody
- Ag:
-
Antigen
- IgG1:
-
Immunoglobulin G1
- CDR:
-
Complementary-determining region
- c-Met:
-
Mesenchymal epithelial transition factor
- VCAM-1:
-
Vascular cell adhesion molecule 1
- DM1:
-
N2’-deacetyl-N2’-(3-mercapto-1-oxopropyl)-maytansine
- HIV:
-
Human immunodeficiency virus
- EGFR:
-
Epidermal growth factor receptor
- MW:
-
Molecular weight
- LAP:
-
Latency-associated peptide
- dsFv:
-
Disulfide-stabilized variable fragment
- H:
-
Heavy chain
- L:
-
Light chain
- MMP:
-
Matrix metalloprotease
- ADAM:
-
A disintegrin and metalloproteinase
- uPA:
-
Urokinase plasminogen activator
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed death-ligand 1
- VEGF-A:
-
Vascular endothelial growth factor A
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- IL:
-
Interleukin
- IL-6R:
-
IL-6 receptor
- RANKL:
-
Receptor activator of nuclear factor kappa-Β ligand
- BACE1:
-
β-site amyloid β precursor protein cleaving enzyme 1
- Aβ:
-
Amyloid β
- mAbs:
-
Monoclonal antibodies
- Pro-Ab:
-
Pro-antibody
- FDA:
-
US Food and Drug Administration
- CAGR:
-
Compound annual growth rate
- RA:
-
Rheumatoid arthritis
- JCV:
-
John Cunningham virus
- NHL:
-
Non-Hodgkin’s lymphoma
- EMA:
-
European Medicines Agency
- CLL:
-
Chronic lymphocytic leukaemia
- CIA:
-
Collagen-induced arthritis
- PK:
-
Pharmacokinetics
- CC:
-
Coiled-coil
- ADCs:
-
Ab-drug conjugates
- aDVD:
-
Activatable dual variable domain
- ICAM-1:
-
Intercellular adhesion molecule 1
- SPR:
-
Surface plasmon resonance
- PSCA:
-
Prostate stem cell antigen
- MT-SP1:
-
Membrane type-serine protease 1
- TMDD:
-
Target-mediated drug disposition
- scFv:
-
Single chain Fv
- sEGFRdIII:
-
Soluble EGFR domain III
- dsDNA:
-
Double strand DNA
- ssDNA:
-
Single strand DNA
- ECM:
-
Extracellular matrix
- ALL:
-
Acute lymphoblastic leukemia
- COPD:
-
Chronic obstructed pulmonary disease
- ADA:
-
Anti-drug antibody
- NF-κB:
-
Nucleus factor kappa B
- n.d.a.:
-
No data available
References
Adenis A, Huet G, Zerimech F, Hecquet B, Balduyck M, Peyrat JP. Cathepsin B, L, and D activities in colorectal carcinomas: relationship with clinico-pathological parameters. Cancer Lett. 1995;96(2):267–75.
Agrawal S, Statkevich P, Bajaj G, Feng Y, Saeger S, Desai DD, Park JS, Waxman IM, Roy A, Gupta M. Evaluation of immunogenicity of Nivolumab monotherapy and its clinical relevance in patients with metastatic solid tumors. J Clin Pharmacol. 2017;57(3):394–400.
Akcilar R, Namdar ND, Kocak FE, Bayat Z, Burhan H. ADAM 33 gene V4 C/G rs2787094 polymorphism in psoriasis. Bratisl Lek Listy. 2018;119(7):454–7.
Aksoy S, Harputluoglu H, Kilickap S, Dede DS, Dizdar O, Altundag K, Barista I. Rituximab-related viral infections in lymphoma patients. Leuk Lymphoma. 2007;48(7):1307–12.
Altrichter S, Boodstein N, Maurer M. Matrix metalloproteinase-9: a novel biomarker for monitoring disease activity in patients with chronic urticaria patients? Allergy. 2009;64(4):652–6.
Anderson DC, Springer TA. Leukocyte adhesion deficiency: an inherited defect in the mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med. 1987;38:175–94.
Anthony DC, Ferguson B, Matyzak MK, Miller KM, Esiri MM, Perry VH. Differential matrix metalloproteinase expression in cases of multiple sclerosis and stroke. Neuropathol Appl Neurobiol. 1997;23(5):406–15.
Askling J, Fored CM, Brandt L, Baecklund E, Bertilsson L, Coster L, Geborek P, Jacobsson LT, Lindblad S, Lysholm J, Rantapaa-Dahlqvist S, Saxne T, Romanus V, Klareskog L, Feltelius N. Risk and case characteristics of tuberculosis in rheumatoid arthritis associated with tumor necrosis factor antagonists in Sweden. Arthritis Rheum. 2005;52(7):1986–92.
Ates A, Turkcapar N, Olmez U, Tiryaki O, Duzgun N, Uguz E, Duman M. Serum pro-matrix metalloproteinase-3 as an indicator of disease activity and severity in rheumatoid arthritis: comparison with traditional markers. Rheumatol Int. 2007;27(8):715–22.
Azevedo A, Prado AF, Feldman S, de Figueiredo FAT, Dos Santos MCG, Issa JPM. MMPs are involved in osteoporosis and are correlated with cardiovascular diseases. Curr Pharm Des. 2018;24(16):1801–10.
Bailon E, Ugarte-Berzal E, Amigo-Jimenez I, Van den Steen P, Opdenakker G, Garcia-Marco JA, Garcia-Pardo A. Overexpression of progelatinase B/proMMP-9 affects migration regulatory pathways and impairs chronic lymphocytic leukemia cell homing to bone marrow and spleen. J Leukoc Biol. 2014;96(2):185–99.
Blanchot-Jossic F, Jarry A, Masson D, Bach-Ngohou K, Paineau J, Denis MG, Laboisse CL, Mosnier JF. Up-regulated expression of ADAM17 in human colon carcinoma: co-expression with EGFR in neoplastic and endothelial cells. J Pathol. 2005;207(2):156–63.
Buggins AG, Mufti GJ, Salisbury J, Codd J, Westwood N, Arno M, Fishlock K, Pagliuca A, Devereux S. Peripheral blood but not tissue dendritic cells express CD52 and are depleted by treatment with alemtuzumab. Blood. 2002;100(5):1715–20.
Buommino E, De Filippis A, Gaudiello F, Balato A, Balato N, Tufano MA, Ayala F. Modification of osteopontin and MMP-9 levels in patients with psoriasis on anti-TNF-alpha therapy. Arch Dermatol Res. 2012;304(6):481–5.
Burkhard P, Stetefeld J, Strelkov SV. Coiled coils: a highly versatile protein folding motif. Trends Cell Biol. 2001;11(2):82–8.
Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11:529–43.
Butkiewicz D, Krzesniak M, Drosik A, Giglok M, Gdowicz-Klosok A, Kosarewicz A, Rusin M, Maslyk B, Gawkowska-Suwinska M, Suwinski R. The VEGFR2, COX-2 and MMP-2 polymorphisms are associated with clinical outcome of patients with inoperable non-small cell lung cancer. Int J Cancer. 2015;137(10):2332–42.
Calabresi PA, Giovannoni G, Confavreux C, Galetta SL, Havrdova E, Hutchinson M, Kappos L, Miller DH, O'Connor PW, Phillips JT, Polman CH, Radue EW, Rudick RA, Stuart WH, Lublin FD, Wajgt A, Weinstock-Guttman B, Wynn DR, Lynn F, Panzara MA, Affirm and Investigators S. The incidence and significance of anti-natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69(14):1391–403.
Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, Furman RR, Vose JM, Zelenetz AD, Mamtani R, Raisch DW, Dorshimer GW, Rosen ST, Muro K, Gottardi-Littell NR, Talley RL, Sartor O, Green D, Major EO, Bennett CL. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113(20):4834–40.
Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, Bennett CL. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the research on adverse drug events and reports (RADAR) project. Lancet Oncol. 2009;10(8):816–24.
Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1(2):118–29.
Casadevall A, Dadachova E, Pirofski LA. Passive antibody therapy for infectious diseases. Nat Rev Microbiol. 2004;2(9):695–703.
Chakrabarti S, MacDonald D, Hale G, Holder K, Turner V, Czarnecka H, Thompson J, Fegan C, Waldmann H, Milligan DW. T-cell depletion with Campath-1H "in the bag" for matched related allogeneic peripheral blood stem cell transplantation is associated with reduced graft-versus-host disease, rapid immune constitution and improved survival. Br J Haematol. 2003;121(1):109–18.
Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10(5):301–16.
Chandran V, Cook RJ, Edwin J, Shen H, Pellett FJ, Shanmugarajah S, Rosen CF, Gladman DD. Soluble biomarkers differentiate patients with psoriatic arthritis from those with psoriasis without arthritis. Rheumatology (Oxford). 2010;49(7):1399–405.
Chen H, Hu Y, Xiang W, Cai Y, Wang Z, Xiao Q, Liu Y, Li Q, Ding K. Prognostic significance of matrix metalloproteinase 7 immunohistochemical expression in colorectal cancer: a meta-analysis. Int J Clin Exp Med. 2015;8(3):3281–90.
Chen IJ, Chuang CH, Hsieh YC, Lu YC, Lin WW, Huang CC, Cheng TC, Cheng YA, Cheng KW, Wang YT, Chen FM, Cheng TL, Tzou SC. Selective antibody activation through protease-activated pro-antibodies that mask binding sites with inhibitory domains. Sci Rep. 2017;7(1):11587.
Chung DE, Kratz F. Development of a novel albumin-binding prodrug that is cleaved by urokinase-type-plasminogen activator (uPA). Bioorg Med Chem Lett. 2006;16(19):5157–63.
Clark IA. The advent of the cytokine storm. Immunol Cell Biol. 2007;85(4):271–3.
Cohen JA, Weiner DB, More KF, Kokai Y, Williams WV, Maguire HC Jr, LiVolsi VA, Greene MI. Expression pattern of the neu (NGL) gene-encoded growth factor receptor protein (p185neu) in normal and transformed epithelial tissues of the digestive tract. Oncogene. 1989;4(1):81–8.
Cossins JA, Clements JM, Ford J, Miller KM, Pigott R, Vos W, Van der Valk P, De Groot CJ. Enhanced expression of MMP-7 and MMP-9 in demyelinating multiple sclerosis lesions. Acta Neuropathol. 1997;94(6):590–8.
Cuzner ML, Opdenakker G. Plasminogen activators and matrix metalloproteases, mediators of extracellular proteolysis in inflammatory demyelination of the central nervous system. J Neuroimmunol. 1999;94(1–2):1–14.
Dass K, Ahmad A, Azmi AS, Sarkar SH, Sarkar FH. Evolving role of uPA/uPAR system in human cancers. Cancer Treat Rev. 2008;34(2):122–36.
Davda J, Declerck P, Hu-Lieskovan S, Hickling TP, Jacobs IA, Chou J, Salek-Ardakani S, Kraynov E. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J Immunother Cancer. 2019;7(1):105.
Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ, Sagert JG, Hostetter DR, Han F, Gee J, Flandez J, Markham K, Nguyen M, Krimm M, Wong KR, Liu S, Daugherty PS, West JW, Lowman HB. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med. 2013;5(207):207ra144.
Donaldson JM, Kari C, Fragoso RC, Rodeck U, Williams JC. Design and development of masked therapeutic antibodies to limit off-target effects: application to anti-EGFR antibodies. Cancer Biol Ther. 2009;8(22):2147–52.
Dong ZL, Lin J, Zhang B, Zhu Y, Li N, Xie S, Wang Y, Gao N, Huang Z. Structural basis of assembly of the human T cell receptor-CD3 complex. Nature. 2019;573(7775):546–52.
Dorweiler B, Torzewski M, Dahm M, Kirkpatrick CJ, Lackner KJ, Vahl CF. Subendothelial infiltration of neutrophil granulocytes and liberation of matrix-destabilizing enzymes in an experimental model of human neo-intima. Thromb Haemost. 2008;99(2):373–81.
Drake MT, Clarke BL, Oursler MJ, Khosla S. Cathepsin K inhibitors for osteoporosis: biology, potential clinical utility, and lessons learned. Endocr Rev. 2017;38(4):325–50.
Duffy MJ, Duggan C, Mulcahy HE, McDermott EW, O'Higgins NJ. Urokinase plasminogen activator: a prognostic marker in breast cancer including patients with axillary node-negative disease. Clin Chem. 1998;44(6 Pt 1):1177–83.
Dziadzio M, Smith R. Meta-analysis is no substitute for a comprehensive national registry. Clin Rheumatol. 2007;26(7):1134–5.
Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. 2015;7(1):9–14.
Erster O, Thomas JM, Hamzah J, Jabaiah AM, Getz JA, Schoep TD, Hall SS, Ruoslahti E, Daugherty PS. Site-specific targeting of antibody activity in vivo mediated by disease-associated proteases. J Control Release. 2012;161(3):804–12.
Feldmann M, Maini SR. Role of cytokines in rheumatoid arthritis: an education in pathophysiology and therapeutics. Immunol Rev. 2008;223:7–19.
Fleischmajer R, Kuroda K, Hazan R, Gordon RE, Lebwohl MG, Sapadin AN, Unda F, Iehara N, Yamada Y. Basement membrane alterations in psoriasis are accompanied by epidermal overexpression of MMP-2 and its inhibitor TIMP-2. J Invest Dermatol. 2000;115(5):771–7.
Foekens JA, Kos J, Peters HA, Krasovec M, Look MP, Cimerman N, Meijer-van Gelder ME, Henzen-Logmans SC, van Putten WL, Klijn JG. Prognostic significance of cathepsins B and L in primary human breast cancer. J Clin Oncol. 1998;16(3):1013–21.
Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008;26(32):5275–83.
Fonovic M, Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta. 2014;1840(8):2560–70.
Galliera E, Tacchini L, Corsi Romanelli MM. Matrix metalloproteinases as biomarkers of disease: updates and new insights. Clin Chem Lab Med. 2015;53(3):349–55.
Gavert N, Conacci-Sorrell M, Gast D, Schneider A, Altevogt P, Brabletz T, Ben-Ze'ev A. L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol. 2005;168(4):633–42.
Gelfand JM, Cree BAC, Hauser SL. Ocrelizumab and other CD20(+) B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics. 2017;14(4):835–41.
Georgescu EF, Mogoanta SS, Costache A, Parvanescu V, Totolici BD, Patrascu S, Stanescu C. The assessment of matrix metalloproteinase-9 expression and angiogenesis in colorectal cancer. Romanian J Morphol Embryol. 2015;56(3):1137–44.
Gerber HP, Kung-Sutherland M, Stone I, Morris-Tilden C, Miyamoto J, McCormick R, Alley SC, Okeley N, Hayes B, Hernandez-Ilizaliturri FJ, McDonagh CF, Carter PJ, Benjamin D, Grewal IS. Potent antitumor activity of the anti-CD19 auristatin antibody drug conjugate hBU12-vcMMAE against rituximab-sensitive and -resistant lymphomas. Blood. 2009;113(18):4352–61.
Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285(5430):1028–32.
Gibbs SD, Westerman DA, McCormack C, Seymour JF, Miles PH. Severe and prolonged myeloid haematopoietic toxicity with myelodysplastic features following alemtuzumab therapy in patients with peripheral T-cell lymphoproliferative disorders. Br J Haematol. 2005;130(1):87–91.
Gijbels K, Masure S, Carton H, Opdenakker G. Gelatinase in the cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological disorders. J Neuroimmunol. 1992;41(1):29–34.
Gormley JA, Hegarty SM, O'Grady A, Stevenson MR, Burden RE, Barrett HL, Scott CJ, Johnston JA, Wilson RH, Kay EW, Johnston PG, Olwill SA. The role of Cathepsin S as a marker of prognosis and predictor of chemotherapy benefit in adjuvant CRC: a pilot study. Br J Cancer. 2011;105(10):1487–94.
Gray E, Thomas TL, Betmouni S, Scolding N, Love S. Elevated matrix metalloproteinase-9 and degradation of perineuronal nets in cerebrocortical multiple sclerosis plaques. J Neuropathol Exp Neurol. 2008;67(9):888–99.
Haider I, Cahill M. Fatal thrombocytopaenia temporally related to the administration of alemtuzumab (MabCampath) for refractory CLL despite early discontinuation of therapy. Hematology. 2004;9(5–6):409–11.
Hale G, Jacobs P, Wood L, Fibbe WE, Barge R, Novitzky N, Toit C, Abrahams L, Thomas V, Bunjes D, Duncker C, Wiesneth M, Selleslag D, Hidajat M, Starobinski M, Bird P, Waldmann H. CD52 antibodies for prevention of graft-versus-host disease and graft rejection following transplantation of allogeneic peripheral blood stem cells. Bone Marrow Transplant. 2000;26(1):69–76.
Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9(4):325–38.
Harbeck N, Alt U, Berger U, Kruger A, Thomssen C, Janicke F, Hofler H, Kates RE, Schmitt M. Prognostic impact of proteolytic factors (urokinase-type plasminogen activator, plasminogen activator inhibitor 1, and cathepsins B, D, and L) in primary breast cancer reflects effects of adjuvant systemic therapy. Clin Cancer Res. 2001;7(9):2757–64.
Hauser SL. Multiple lessons for multiple sclerosis. N Engl J Med. 2008;359(17):1838–41.
Hsu DW, Efird JT, Hedley-Whyte ET. Prognostic role of urokinase-type plasminogen activator in human gliomas. Am J Pathol. 1995;147(1):114–23.
Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol Cell Biol. 1989;9(3):1165–72.
Inghirami G, Wieczorek R, Zhu BY, Silber R, Dalla-Favera R, Knowles DM. Differential expression of LFA-1 molecules in non-Hodgkin's lymphoma and lymphoid leukemia. Blood. 1988;72(4):1431–4.
Investigators CT, Coles AJ, Compston DA, Selmaj KW, Lake SL, Moran S, Margolin DH, Norris K, Tandon PK. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359(17):1786–801.
Ishikawa N, Daigo Y, Yasui W, Inai K, Nishimura H, Tsuchiya E, Kohno N, Nakamura Y. ADAM8 as a novel serological and histochemical marker for lung cancer. Clin Cancer Res. 2004;10(24):8363–70.
Izycka-Swieszewska E, Wozniak A, Drozynska E, Kot J, Grajkowska W, Klepacka T, Perek D, Koltan S, Bien E, Limon J. Expression and significance of HER family receptors in neuroblastic tumors. Clin Exp Metastasis. 2011;28(3):271–82.
Janssen BMG, Lempens EHM, Olijve LLC, Voets IK, van Dongen JLJ, de Greef TFA, Merkx M. Reversible blocking of antibodies using bivalent peptide-DNA conjugates allows protease-activatable targeting. Chem Sci. 2013;4:1442–50.
Ji F, Chen YL, Jin EY, Wang WL, Yang ZL, Li YM. Relationship between matrix metalloproteinase-2 mRNA expression and clinicopathological and urokinase-type plasminogen activator system parameters and prognosis in human gastric cancer. World J Gastroenterol. 2005;11(21):3222–6.
Jiang T, Olson ES, Nguyen QT, Roy M, Jennings PA, Tsien RY. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101(51):17867–72.
Karaca M, Kocoglu H, Bilgetekin I, Ozet A, Sahinli H, Demir H, Kankoc A, Tural D, Yucel OK. Ventricular bigeminal rhythm associated with trastuzumab: A potential cardiac side effect. J Cancer Res Ther. 2018;14(Supplement):S536–7.
Kaufman HL, Wolchok JD. Is tumor immunity the same thing as autoimmunity? Implications for cancer immunotherapy. J Clin Oncol. 2006;24(15):2230–2.
Kawaguchi M, Mitsuhashi Y, Kondo S. Overexpression of tumour necrosis factor-alpha-converting enzyme in psoriasis. Br J Dermatol. 2005;152(5):915–9.
Kayser K, Richter N, Hufnagl P, Kayser G, Kos J, Werle B. Expression, proliferation activity and clinical significance of cathepsin B and cathepsin L in operated lung cancer. Anticancer Res. 2003;23(3C):2767–72.
Keane J. TNF-blocking agents and tuberculosis: new drugs illuminate an old topic. Rheumatology (Oxford). 2005;44(6):714–20.
Keefe DL. Trastuzumab-associated cardiotoxicity. Cancer. 2002;95(7):1592–600.
Kenter MJ, Cohen AF. Establishing risk of human experimentation with drugs: lessons from TGN1412. Lancet. 2006;368(9544):1387–91.
King KM, Younes A. Rituximab: review and clinical applications focusing on non-Hodgkin's lymphoma. Expert Rev Anticancer Ther. 2001;1(2):177–86.
Klotz L, Havla J, Schwab N, Hohlfeld R, Barnett M, Reddel S, Wiendl H. Risks and risk management in modern multiple sclerosis immunotherapeutic treatment. Ther Adv Neurol Disord. 2019;12:1756286419836571.
Kobayashi H, Fujishiro S, Terao T. Impact of urokinase-type plasminogen activator and its inhibitor type 1 on prognosis in cervical cancer of the uterus. Cancer Res. 1994;54(24):6539–48.
Kos J, Sekirnik A, Kopitar G, Cimerman N, Kayser K, Stremmer A, Fiehn W, Werle B. Cathepsin S in tumours, regional lymph nodes and sera of patients with lung cancer: relation to prognosis. Br J Cancer. 2001;85(8):1193–200.
Lah TT, Cercek M, Blejec A, Kos J, Gorodetsky E, Somers R, Daskal I. Cathepsin B, a prognostic indicator in lymph node-negative breast carcinoma patients: comparison with cathepsin D, cathepsin L, and other clinical indicators. Clin Cancer Res. 2000;6(2):578–84.
Lehvaslaiho H, Lehtola L, Sistonen L, Alitalo K. A chimeric EGF-R-neu proto-oncogene allows EGF to regulate neu tyrosine kinase and cell transformation. EMBO J. 1989;8(1):159–66.
Lendeckel U, Kohl J, Arndt M, Carl-McGrath S, Donat H, Rocken C. Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. J Cancer Res Clin Oncol. 2005;131(1):41–8.
Lenglet S, Mach F, Montecucco F. Role of matrix metalloproteinase-8 in atherosclerosis. Mediat Inflamm. 2013;2013:659282.
Leo O, Foo M, Sachs DH, Samelson LE, Bluestone JA. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci U S A. 1987;84(5):1374–8.
Leppert D, Ford J, Stabler G, Grygar C, Lienert C, Huber S, Miller KM, Hauser SL, Kappos L. Matrix metalloproteinase-9 (gelatinase B) is selectively elevated in CSF during relapses and stable phases of multiple sclerosis. Brain. 1998;121(Pt 12):2327–34.
Li H, Ye X, Mahanivong C, Bian D, Chun J, Huang S. Signaling mechanisms responsible for lysophosphatidic acid-induced urokinase plasminogen activator expression in ovarian cancer cells. J Biol Chem. 2005;280(11):10564–71.
Lin TS. Novel agents in chronic lymphocytic leukemia: efficacy and tolerability of new therapies. Clin Lymphoma Myeloma. 2008;8(Suppl 4):S137–43.
Lindberg RL, De Groot CJ, Montagne L, Freitag P, van der Valk P, Kappos L, Leppert D. The expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in lesions and normal appearing white matter of multiple sclerosis. Brain. 2001;124(Pt 9):1743–53.
Liu B, Guo H, Zhang J, Xue J, Yang Y, Qin T, Xu J, Guo Q, Zhang D, Qian W, Li B, Hou S, Dai J, Guo Y, Wang H. In-depth characterization of a pro-antibody-drug conjugate by LC-MS. Mol Pharm. 2016;13(8):2702–10.
Liu B, Spearman M, Doering J, Lattova E, Perreault H, Butler M. The availability of glucose to CHO cells affects the intracellular lipid-linked oligosaccharide distribution, site occupancy and the N-glycosylation profile of a monoclonal antibody. J Biotechnol. 2014;170:17–27.
Loertscher R. The utility of monoclonal antibody therapy in renal transplantation. Transplant Proc. 2002;34(3):797–800.
Look MP, van Putten WL, Duffy MJ, Harbeck N, Christensen IJ, Thomssen C, Kates R, Spyratos F, Ferno M, Eppenberger-Castori S, Sweep CG, Ulm K, Peyrat JP, Martin PM, Magdelenat H, Brunner N, Duggan C, Lisboa BW, Bendahl PO, Quillien V, Daver A, Ricolleau G, Meijer-van Gelder ME, Manders P, Fiets WE, Blankenstein MA, Broet P, Romain S, Daxenbichler G, Windbichler G, Cufer T, Borstnar S, Kueng W, Beex LV, Klijn JG, O'Higgins N, Eppenberger U, Janicke F, Schmitt M, Foekens JA. Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J Natl Cancer Inst. 2002;94(2):116–28.
Lopez-Diego RS, Weiner HL. Novel therapeutic strategies for multiple sclerosis--a multifaceted adversary. Nat Rev Drug Discov. 2008;7(11):909–25.
Lu F, Toh PC, Burnett I, Li F, Hudson T, Amanullah A, Li J. Automated dynamic fed-batch process and media optimization for high productivity cell culture process development. Biotechnol Bioeng. 2013;110(1):191–205.
Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, Wu HC. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020;27(1):1.
Lu YC, Chuang CH, Chuang KH, Chen IJ, Huang BC, Lee WH, Wang HE, Li JJ, Cheng YA, Cheng KW, Wang JY, Hsieh YC, Lin WW, Cheng TL. Specific activation of pro-infliximab enhances selectivity and safety of rheumatoid arthritis therapy. PLoS Biol. 2019;17(6):e3000286.
Lutterotti A, Martin R. Getting specific: monoclonal antibodies in multiple sclerosis. Lancet Neurol. 2008;7(6):538–47.
Maguire HC Jr, Greene MI. The neu (c-erbB-2) oncogene. Semin Oncol. 1989;16(2):148–55.
Maker AV, Attia P, Rosenberg SA. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J Immunol. 2005;175(11):7746–54.
Matusiewicz M, Neubauer K, Mierzchala-Pasierb M, Gamian A, Krzystek-Korpacka M. Matrix metalloproteinase-9: its interplay with angiogenic factors in inflammatory bowel diseases. Dis Markers. 2014;2014:643645.
Maximiano S, Magalhaes P, Guerreiro MP, Morgado M. Trastuzumab in the treatment of breast Cancer. BioDrugs. 2016;30(2):75–86.
McClain DL, Woods HL, Oakley MG. Design and characterization of a heterodimeric coiled coil that forms exclusively with an antiparallel relative helix orientation. J Am Chem Soc. 2001;123(13):3151–2.
McGowan PM, McKiernan E, Bolster F, Ryan BM, Hill AD, McDermott EW, Evoy D, O'Higgins N, Crown J, Duffy MJ. ADAM-17 predicts adverse outcome in patients with breast cancer. Ann Oncol. 2008;19(6):1075–81.
Metz S, Panke C, Haas AK, Schanzer J, Lau W, Croasdale R, Hoffmann E, Schneider B, Auer J, Gassner C, Bossenmaier B, Umana P, Sustmann C, Brinkmann U. Bispecific antibody derivatives with restricted binding functionalities that are activated by proteolytic processing. Protein Eng Des Sel. 2012;25(10):571–80.
Mezentsev A, Nikolaev A, Bruskin S. Matrix metalloproteinases and their role in psoriasis. Gene. 2014;540(1):1–10.
Miranda O, Farooqui M, Siegfried JM. Status of Agents Targeting the HGF/c-Met Axis in Lung Cancer. Cancers (Basel). 2018;10(9).
Mitoma H, Horiuchi T, Tsukamoto H, Ueda N. Molecular mechanisms of action of anti-TNF-alpha agents - comparison among therapeutic TNF-alpha antagonists. Cytokine. 2018;101:56–63.
Mitsui Y, Mochizuki S, Kodama T, Shimoda M, Ohtsuka T, Shiomi T, Chijiiwa M, Ikeda T, Kitajima M, Okada Y. ADAM28 is overexpressed in human breast carcinomas: implications for carcinoma cell proliferation through cleavage of insulin-like growth factor binding protein-3. Cancer Res. 2006;66(20):9913–20.
Miyasaka N, Investigators CS. Clinical investigation in highly disease-affected rheumatoid arthritis patients in Japan with adalimumab applying standard and general evaluation: the CHANGE study. Mod Rheumatol. 2008;18(3):252–62.
Molloy ES, Calabrese LH. Therapy: targeted but not trouble-free: efalizumab and PML. Nat Rev Rheumatol. 2009;5(8):418–9.
Moss ML, Sklair-Tavron L, Nudelman R. Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2008;4(6):300–9.
Mukohara T. Role of HER2-targeted agents in adjuvant treatment for breast Cancer. Chemother Res Pract. 2011;2011:730360.
Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer. 2008;8(12):929–41.
Netzel-Arnett S, Sang QX, Moore WG, Navre M, Birkedal-Hansen H, Van Wart HE. Comparative sequence specificities of human 72- and 92-kDa gelatinases (type IV collagenases) and PUMP (matrilysin). Biochemistry. 1993;32(25):6427–32.
Nielsen A, Scarlett CJ, Samra JS, Gill A, Li Y, Allen BJ, Smith RC. Significant overexpression of urokinase-type plasminogen activator in pancreatic adenocarcinoma using real-time quantitative reverse transcription polymerase chain reaction. J Gastroenterol Hepatol. 2005;20(2):256–63.
Nouh MA, Mohamed MM, El-Shinawi M, Shaalan MA, Cavallo-Medved D, Khaled HM, Sloane BF. Cathepsin B: a potential prognostic marker for inflammatory breast cancer. J Transl Med. 2011;9:1.
O'Shea C, McKie N, Buggy Y, Duggan C, Hill AD, McDermott E, O'Higgins N, Duffy MJ. Expression of ADAM-9 mRNA and protein in human breast cancer. Int J Cancer. 2003;105(6):754–61.
Ohtsuka T, Shiomi T, Shimoda M, Kodama T, Amour A, Murphy G, Ohuchi E, Kobayashi K, Okada Y. ADAM28 is overexpressed in human non-small cell lung carcinomas and correlates with cell proliferation and lymph node metastasis. Int J Cancer. 2006;118(2):263–73.
Olson OC, Joyce JA. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat Rev Cancer. 2015;15(12):712–29.
Onuoha SC, Ferrari M, Sblattero D, Pitzalis C. Rational design of antirheumatic prodrugs specific for sites of inflammation. Arthritis Rheum. 2015;67(10):2661–72.
Pai CS, Simons DM, Lu X, Evans M, Wei J, Wang YH, Chen M, Huang J, Park C, Chang A, Wang J, Westmoreland S, Beam C, Banach D, Bowley D, Dong F, Seagal J, Ritacco W, Richardson PL, Mitra S, Lynch G, Bousquet P, Mankovich J, Kingsbury G, Fong L. Tumor-conditional anti-CTLA4 uncouples antitumor efficacy from immunotherapy-related toxicity. J Clin Invest. 2019;129(1):349–63.
Paik S, Tang G, Shak S, Kim C, Baker J, Kim W, Cronin M, Baehner FL, Watson D, Bryant J, Costantino JP, Geyer CE Jr, Wickerham DL, Wolmark N. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol. 2006;24(23):3726–34.
Pedersen G, Saermark T, Kirkegaard T, Brynskov J. Spontaneous and cytokine induced expression and activity of matrix metalloproteinases in human colonic epithelium. Clin Exp Immunol. 2009;155(2):257–65.
Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006;18(2):206–13.
Perez EA. Cardiac toxicity of ErbB2-targeted therapies: what do we know? Clin Breast Cancer. 2008;8(Suppl 3):S114–20.
Plevy S, Salzberg B, Van Assche G, Regueiro M, Hommes D, Sandborn W, Hanauer S, Targan S, Mayer L, Mahadevan U, Frankel M, Lowder J. A phase I study of visilizumab, a humanized anti-CD3 monoclonal antibody, in severe steroid-refractory ulcerative colitis. Gastroenterology. 2007;133(5):1414–22.
Pluckthun A, Pack P. New protein engineering approaches to multivalent and bispecific antibody fragments. Immunotechnology. 1997;3(2):83–105.
Povey JF, O'Malley CJ, Root T, Martin EB, Montague GA, Feary M, Trim C, Lang DA, Alldread R, Racher AJ, Smales CM. Rapid high-throughput characterisation, classification and selection of recombinant mammalian cell line phenotypes using intact cell MALDI-ToF mass spectrometry fingerprinting and PLS-DA modelling. J Biotechnol. 2014;184:84–93.
Poyer F, Coquerel B, Pegahi R, Cazin L, Norris V, Vannier JP, Lamacz M. Secretion of MMP-2 and MMP-9 induced by VEGF autocrine loop correlates with clinical features in childhood acute lymphoblastic leukemia. Leuk Res. 2009;33(3):407–17.
Pulukuri SM, Gondi CS, Lakka SS, Jutla A, Estes N, Gujrati M, Rao JS. RNA interference-directed knockdown of urokinase plasminogen activator and urokinase plasminogen activator receptor inhibits prostate cancer cell invasion, survival, and tumorigenicity in vivo. J Biol Chem. 2005;280(43):36529–40.
Ransohoff RM. Natalizumab for multiple sclerosis. N Engl J Med. 2007;356(25):2622–9.
Rath T, Roderfeld M, Halwe JM, Tschuschner A, Roeb E, Graf J. Cellular sources of MMP-7, MMP-13 and MMP-28 in ulcerative colitis. Scand J Gastroenterol. 2010;45(10):1186–96.
Ratzinger G, Reagan JL, Heller G, Busam KJ, Young JW. Differential CD52 expression by distinct myeloid dendritic cell subsets: implications for alemtuzumab activity at the level of antigen presentation in allogeneic graft-host interactions in transplantation. Blood. 2003;101(4):1422–9.
Ren F, Tang R, Zhang X, Madushi WM, Luo D, Dang Y, Li Z, Wei K, Chen G. Overexpression of MMP family members functions as prognostic biomarker for breast Cancer patients: a systematic review and meta-analysis. PLoS One. 2015;10(8):e0135544.
Rogliani P, Calzetta L, Ora J, Matera MG. Canakinumab for the treatment of chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2015;31:15–27.
Rosh JR, Gross T, Mamula P, Griffiths A, Hyams J. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn's disease: a cautionary tale? Inflamm Bowel Dis. 2007;13(8):1024–30.
Rossetti C, Reis BC, Delgado PO, Azzalis LA, Junqueira VB, Feder D, Fonseca F. Adhesion molecules in breast carcinoma: a challenge to the pathologist. Rev Assoc Med Bras. 1992;61(1):81 -85, 2015.
Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, Wiggins J, Barnes PJ. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26(5):602–9.
Sadiq SA, Puccio LM, Brydon EW. JCV detection in multiple sclerosis patients treated with natalizumab. J Neurol. 2010;257(6):954–8.
Salaun M, Peng J, Hensley HH, Roder N, Flieder DB, Houlle-Crepin S, Abramovici-Roels O, Sabourin JC, Thiberville L, Clapper ML. MMP-13 in-vivo molecular imaging reveals early expression in lung adenocarcinoma. PLoS One. 2015;10(7):e0132960.
Scheinfeld N. Adalimumab: a review of side effects. Expert Opin Drug Saf. 2005;4(4):637–41.
Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. J Dermatological Treatment. 2004;15(5):280–94.
Scheinfeld N. Efalizumab: a review of events reported during clinical trials and side effects. Expert Opin Drug Saf. 2006;5(2):197–209.
Scherrer A, Wohlwend A, Kruithof EK, Vassalli JD, Sappino AP. Plasminogen activation in human acute leukaemias. Br J Haematol. 1999;105(4):920–7.
Schmidt MM. Engineering antibodies for improved targeting of solid tumors. PhD thesis, Massachusetts Institute of Technology:Ch. 5; 2010.
Schutz A, Hartig W, Wobus M, Grosche J, Wittekind C, Aust G. Expression of ADAM15 in lung carcinomas. Virchows Arch. 2005;446(4):421–9.
Schweiger A, Staib A, Werle B, Krasovec M, Lah TT, Ebert W, Turk V, Kos J. Cysteine proteinase cathepsin H in tumours and sera of lung cancer patients: relation to prognosis and cigarette smoking. Br J Cancer. 2000;82(4):782–8.
Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12(4):278–87.
Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, Brogi E, Brastianos PK, Hahn WC, Holsinger LJ, Massague J, Leslie CS, Joyce JA. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. 2014;16(9):876–88.
Sgro C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: bibliographic review. Toxicology. 1995;105(1):23–9.
Shapiro SD. Elastolytic metalloproteinases produced by human mononuclear phagocytes. Potential roles in destructive lung disease. Am J Respir Crit Care Med. 1994;150(6 Pt 2):S160–4.
Shiomi H, Eguchi Y, Tani T, Kodama M, Hattori T. Cellular distribution and clinical value of urokinase-type plasminogen activator, its receptor, and plasminogen activator inhibitor-2 in esophageal squamous cell carcinoma. Am J Pathol. 2000;156(2):567–75.
Simonetti O, Lucarini G, Goteri G, Zizzi A, Biagini G, Lo ML, Offidani A. VEGF is likely a key factor in the link between inflammation and angiogenesis in psoriasis: results of an immunohistochemical study. Int J Immunopathol Pharmacol. 2006;19(4):751–60.
Sloane BF. Cathepsin B and cystatins: evidence for a role in cancer progression. Semin Cancer Biol. 1990;1(2):137–52.
Smolarz B, Blasiak J, Kulig A, Romanowicz-Makowska H, Dziki A, Ulanska J, Pander B, Szewczyk T. Plasminogen activator inhibitor 1 (PAI-1) levels and gene promoter polymorphisms in subjects with colorectal cancer. J Exp Clin Cancer Res. 2001;20(2):247–52.
Starodubtseva NL, Sobolev VV, Soboleva AG, Nikolaev AA, Bruskin SA. Expression of genes for metalloproteinases (MMP-1, MMP-2, MMP-9, and MMP-12) associated with psoriasis. Genetika. 2011;47(9):1254–61.
Stern DF, Kamps MP. EGF-stimulated tyrosine phosphorylation of p185neu: a potential model for receptor interactions. EMBO J. 1988;7(4):995–1001.
Stillfried GE, Saunders DN, Ranson M. Plasminogen binding and activation at the breast cancer cell surface: the integral role of urokinase activity. Breast Cancer Res. 2007;9(1):R14.
Sukoh N, Abe S, Ogura S, Isobe H, Takekawa H, Inoue K, Kawakami Y. Immunohistochemical study of cathepsin B. Prognostic significance in human lung cancer. Cancer. 1994;74(1):46–51.
Sun S, Bay-Jensen AC, Karsdal MA, Siebuhr AS, Zheng Q, Maksymowych WP, Christiansen TG, Henriksen K. The active form of MMP-3 is a marker of synovial inflammation and cartilage turnover in inflammatory joint diseases. BMC Musculoskelet Disord. 2014;15:93.
Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–28.
Suomela S, Kariniemi AL, Impola U, Karvonen SL, Snellman E, Uurasmaa T, Peltonen J, Saarialho-Kere U. Matrix metalloproteinase-19 is expressed by keratinocytes in psoriasis. Acta Derm Venereol. 2003;83(2):108–14.
Suomela S, Kariniemi AL, Snellman E, Saarialho-Kere U. Metalloelastase (MMP-12) and 92-kDa gelatinase (MMP-9) as well as their inhibitors, TIMP-1 and -3, are expressed in psoriatic lesions. Exp Dermatol. 2001;10(3):175–83.
Swiercz R, Wolfe JD, Zaher A, Jankun J. Expression of the plasminogen activation system in kidney cancer correlates with its aggressive phenotype. Clin Cancer Res. 1998;4(4):869–77.
Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017;18(12):e731–41.
Tan-Chiu E, Yothers G, Romond E, Geyer CE Jr, Ewer M, Keefe D, Shannon RP, Swain SM, Brown A, Fehrenbacher L, Vogel VG, Seay TE, Rastogi P, Mamounas EP, Wolmark N, Bryant J. Assessment of cardiac dysfunction in a randomized trial comparing doxorubicin and cyclophosphamide followed by paclitaxel, with or without trastuzumab as adjuvant therapy in node-positive, human epidermal growth factor receptor 2-overexpressing breast cancer: NSABP B-31. J Clin Oncol. 2005;23(31):7811–9.
Tavakolpour S, Alesaeidi S, Darvishi M, GhasemiAdl M, Darabi-Monadi S, Akhlaghdoust M, Elikaei BS, Jafarieh A. A comprehensive review of rituximab therapy in rheumatoid arthritis patients. Clin Rheumatol. 2019;38(11):2977–94.
Taylor PC, Feldmann M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):578–82.
Thomas F, Boyle AL, Burton AJ, Woolfson DN. A set of de novo designed parallel heterodimeric coiled coils with quantified dissociation constants in the micromolar to sub-nanomolar regime. J Am Chem Soc. 2013;135(13):5161–6.
Thomssen C, Schmitt M, Goretzki L, Oppelt P, Pache L, Dettmar P, Janicke F, Graeff H. Prognostic value of the cysteine proteases cathepsins B and cathepsin L in human breast cancer. Clin Cancer Res. 1995;1(7):741–6.
Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117(2):244–79.
Trang VH, Zhang X, Yumul RC, Zeng W, Stone IJ, Wo SW, Dominguez MM, Cochran JH, Simmons JK, Ryan MC, Lyon RP, Senter PD, Levengood MR. A coiled-coil masking domain for selective activation of therapeutic antibodies. Nat Biotechnol. 2019;37(7):761–5.
Troy AM, Sheahan K, Mulcahy HE, Duffy MJ, Hyland JM, O'Donoghue DP. Expression of Cathepsin B and L antigen and activity is associated with early colorectal cancer progression. Eur J Cancer. 2004;40(10):1610–6.
Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol. 2001;19(7):661–7.
van de Putte LB, Atkins C, Malaise M, Sany J, Russell AS, van Riel PL, Settas L, Bijlsma JW, Todesco S, Dougados M, Nash P, Emery P, Walter N, Kaul M, Fischkoff S, Kupper H. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63(5):508–16.
van Schouwenburg PA, van de Stadt LA, de Jong RN, van Buren EE, Kruithof S, de Groot E, Hart M, van Ham SM, Rispens T, Aarden L, Wolbink GJ, Wouters D. Adalimumab elicits a restricted anti-idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann Rheum Dis. 2013;72(1):104–9.
Vargas DL, Tyor WR. Update on disease-modifying therapies for multiple sclerosis. J Investig Med. 2017;65(5):883–91.
Watson CJ, Bradley JA, Friend PJ, Firth J, Taylor CJ, Bradley JR, Smith KG, Thiru S, Jamieson NV, Hale G, Waldmann H, Calne R. Alemtuzumab (CAMPATH 1H) induction therapy in cadaveric kidney transplantation--efficacy and safety at five years. Am J Transplant. 2005;5(6):1347–53.
Weber J. Review: anti-CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12(7):864–72.
Werle B, Lotterle H, Schanzenbacher U, Lah TT, Kalman E, Kayser K, Bulzebruck H, Schirren J, Krasovec M, Kos J, Spiess E. Immunochemical analysis of cathepsin B in lung tumours: an independent prognostic factor for squamous cell carcinoma patients. Br J Cancer. 1999;81(3):510–9.
Wing M. Monoclonal antibody first dose cytokine release syndromes-mechanisms and prediction. J Immunotoxicol. 2008;5(1):11–5.
Wing MG, Moreau T, Greenwood J, Smith RM, Hale G, Isaacs J, Waldmann H, Lachmann PJ, Compston A. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J Clin Invest. 1996;98(12):2819–26.
Wing MG, Waldmann H, Isaacs J, Compston DA, Hale G. Ex-vivo whole blood cultures for predicting cytokine-release syndrome: dependence on target antigen and antibody isotype. Ther Immunol. 1995;2(4):183–90.
Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood. 1999;94(7):2217–24.
Yan Q, Yuan Y, Yankui L, Jingjie F, Linfang J, Yong P, Dong H, Xiaowei Q. The expression and significance of CXCR5 and MMP-13 in colorectal Cancer. Cell Biochem Biophys. 2015;73(1):253–9.
Yan R, Vassar R. Targeting the beta secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 2014;13(3):319–29.
Yang WC, Lu J, Kwiatkowski C, Yuan H, Kshirsagar R, Ryll T, Huang YM. Perfusion seed cultures improve biopharmaceutical fed-batch production capacity and product quality. Biotechnol Prog. 2014;30(3):616–25.
Yang Y, Guo Q, Chen X, Zhang J, Guo H, Qian W, Hou S, Dai J, Li B, Guo Y, Wang H. Preclinical studies of a pro-antibody-drug conjugate designed to selectively target EGFR-overexpressing tumors with improved therapeutic efficacy. MAbs. 2016;8(2):405–13.
Yang Y, Guo Q, Xia M, Li Y, Peng X, Liu T, Tong X, Xu J, Guo H, Qian W, Hou S, Dai J, Wang H, Liu R, Guo Y. Generation and characterization of a target-selectively activated antibody against epidermal growth factor receptor with enhanced anti-tumor potency. MAbs. 2015;7(2):440–50.
Young-Min S, Cawston T, Marshall N, Coady D, Christgau S, Saxne T, Robins S, Griffiths I. Biomarkers predict radiographic progression in early rheumatoid arthritis and perform well compared with traditional markers. Arthritis Rheum. 2007;56(10):3236–47.
Yousry TA, Major EO, Ryschkewitsch C, Fahle G, Fischer S, Hou J, Curfman B, Miszkiel K, Mueller-Lenke N, Sanchez E, Barkhof F, Radue EW, Jager HR, Clifford DB. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med. 2006;354(9):924–33.
Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, Nograles KE, Guttman-Yassky E, Cardinale I, Lowes MA, Krueger JG. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124(5):1022–10 e1021–1395.
Zheng CG, Chen R, Xie JB, Liu CB, Jin Z, Jin C. Immunohistochemical expression of Notch1, Jagged1, NF-kappaB and MMP-9 in colorectal cancer patients and the relationship to clinicopathological parameters. Cancer Biomark. 2015;15(6):889–97.
Zhou J, Sun D, Xu L, Sun L, Fu S, Li Y. ADAM33 as a psoriasis susceptibility gene in the Han population of northeastern China. Dermatology. 2011;223(4):356–62.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Ministry of Science and Technology, Taipei, Taiwan (MOST 107-2320-B-037-024 -MY3, MOST107-2320-B-037-028-MY2 and MOST106-2311-B-037-001-MY2); the National Health Research Institutes, Taiwan (NHRI-EX107-10729EI and NHRI-EX108-10729EI); Academia Sinica, Taiwan (AS-107-TP-B11 and AS-TP-107-L11); the Program for Translational Innovation of Biopharmaceutical Development-Technology Supporting Platform Axis, Academia Sinica, Taiwan (Grant No. AS-KPQ-106-TSPA); the Ministry of Education, Taiwan (108RSB0029); the KMU-KMUH Co-Project of Key Research (KMU-DK108002 and KMU-DK109001) and Research Foundation (KMU-Q108001, KMU-Q109002 and KMU-DK109004) from Kaohsiung Medical University, Taiwan; the CCH-KMU joint research project (108-CCH-KMU-010); the NSYSU-KMU joint research project (NK-108I08). We also thank the Drug Development and Value Creation Research Center, Kaohsiung Medical University, Taiwan (KMU-TC108A03) for the instrumentation and equipment support.
Author information
Authors and Affiliations
Contributions
Wen-Wei Lin, Yun-Chi Lu and Tian-Lu Cheng wrote the manuscript. Wen-Wei Lin, Chih-Hung Chuang and Tian-Lu Cheng designed and illustrated Tables. Wen-Wei Lin and Tian-Lu Cheng designed and illustrated Figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lin, WW., Lu, YC., Chuang, CH. et al. Ab locks for improving the selectivity and safety of antibody drugs. J Biomed Sci 27, 76 (2020). https://doi.org/10.1186/s12929-020-00652-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-020-00652-z