
Abstract
Immune checkpoints or coinhibitory receptors, such as cytotoxic T lymphocyte antigen (CTLA)-4 and programmed death (PD)-1, play important roles in regulating T cell responses, and they were proven to be effective targets in treating cancer. In chronic viral infections and cancer, T cells are chronically exposed to persistent antigen stimulation. This is often associated with deterioration of T cell function with constitutive activation of immune checkpoints, a state called ‘exhaustion’, which is commonly associated with inefficient control of tumors and persistent viral infections. Immune checkpoint blockade can reinvigorate dysfunctional/exhausted T cells by restoring immunity to eliminate cancer or virus-infected cells. These immune checkpoint blocking antibodies have moved immunotherapy into a new era, and they represent paradigm-shifting therapeutic strategies for cancer treatment. A clearer understanding of the regulatory roles of these receptors and elucidation of the mechanisms of T cell dysfunction will provide more insights for rational design and development of cancer therapies that target immune checkpoints. This article reviews recent advance(s) in molecular understanding of T cell dysfunction in tumor microenvironments. In addition, we also discuss new immune checkpoint targets in cancer therapy.
Similar content being viewed by others
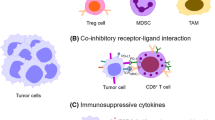


Background
Cancer evades antitumor immune attacks via both inhibiting recognition of cancer specific antigens by T cells and causing dysfunction of CD8 cytotoxic T cells (CTL). Recent breakthroughs and encouraging clinical results with various immune checkpoint inhibitors, such as anti-PD-1 monoclonal antibodies (mAbs) and anti-CTLA-4 mAbs, have demonstrated tremendous potential to control cancer by immune activation [1,2,3,4,5,6,7,8,9]. Immune checkpoint blockade is able to reinvigorate dysfunctional/exhausted T cells by restoring tumor-specific immunity to eliminate cancer cells. In addition to melanoma, inspiring results were reported in other cancers including lung cancer, renal cell carcinoma, bladder cancer, and additional approvals are expected, indicating the great promise held by these mAbs. All these results clearly indicate that a new era of immunotherapy has arrived. Long-term control of cancer with durable treatment response now seems achievable. These mAbs have added a new cornerstone to immunotherapy, making it,another key pillar for cancer treatment in the near future. Immune checkpoint blockade has greatly expanded our knowledge of antitumor immunity and has introduced radical changes and new trends in cancer therapy. Moreover, multiple new immune checkpoints that represent potential new targets for cancer therapy are now under active development. This article reviews advance(s) in recent molecular understanding of T cell dysfunction within tumor microenvironments and of developments of new immune checkpoint therapeutic targets for cancer.
Immune checkpoints or coinhibitory receptors play critical roles in immune homeostasis
To eradicate tumor cells and induce antitumor immunity, T cells are able to recognize tumor antigens presented to T cell receptors (TCRs) by antigen-presenting cells (APCs). After binding to TCR, a second signal (signal two, also called costimulatory signal) is needed for T cell activation. The costimulatory signal comes from the binding of CD28 molecule on T cells with its ligand, B-7 molecules (CD80 and CD86) on APCs. CTLA-4, an immune checkpoint or coinhibitory receptor is induced after T cell activation. CTLA-4 has a higher binding affinity for B-7 ligands than CD28, and CTLA-4 can bind to B7 and displace CD28, leading to attenuation and termination of T cell responses and establishment of tolerance, to minimize the development of autoimmunity. Immune checkpoints or coinhibitory receptors have a central role in regulating autoimmunity, and deficiency of CTLA-4 develops profound lymphoproliferation and systemic autoimmune disease [10, 11]. PD-1 pathway was recognized to play a regulatory role in inhibiting T cell activation and restraining T cell function [12, 13], and PD-1 knockout mice developed proliferative arthritis and a lupus-like autoimmune diseases [14]. Many checkpoint receptors have been genetically associated with autoimmunity and inflammatory diseases [15,16,17,18], suggesting that immune checkpoints or coinhibitory receptors play a critical role in immune tolerance and regulating homeostasis. Therefore, immune checkpoints in regulating T cell activation and immune tolerance have been widely studied. More recently, a new frontier in anticancer [6, 19,20,21] and antiviral therapy [22] has emerged, in which these receptors are being targeted to improve T cell responses [23,24,25].
CTLA-4 as a coinhibitory receptor for T cell activation
The process of T cell activation is tightly regulated by costimulatory signals for full activation, and it is also regulated by coinhibitory signals [26]. The main costimulatory signals for T cell activation are from the B7-1 or B7-2 molecules on antigen presenting cells, which can bind to CD28 on T cells. After binding to its specific antigen ligand, the resulting TCR signals in conjunction with the costimulatory signals from CD28/B7 interaction lead to fully activation of T cells and production of cytokines [27]. CTLA-4 is a major coinhibitory receptor in regulation of T cell response during the priming phase [28]. In contrast to CD28, CTLA-4 delivers an inhibitory signal, and it has a much higher affinity for B7 than CD28 [29, 30]. Thus, CTLA-4 competes for binding to B7, and thereby prevents CD28-mediated T cell costimulation, and also inhibits T cell activation [29, 31, 32]. Moreover, CTLA-4 can capture B7, which induces the degradation of these ligands within the cell via trans-endocytosis [33]. All these effects dampen T cell activation and enhance immune tolerance. In addition, CTLA-4 is essential for the regulatory T cells (Tregs) function [34, 35]. Tregs require CTLA-4 to maintain their function to suppress immune responses, and deficiency of CTLA-4 results in development of profound systemic autoimmune diseases [10, 11]. The concept of using immune checkpoint inhibitors to break T cell dysfunction in tumor patients appears to be an intriguing approach in cancer therapy. This was first demonstrated by the success of Ipilimumab, an anti-CTLA-4 mAb, resulting in the approval of Ipilimumab by the FDA for advanced melanoma [2]. All these results indicate a major conceptual breakthrough in cancer immunotherapy. Immune checkpoint blockade is game-changing and revolutionary in at least in two senses. First, the target for therapy is on immune cells but not tumor cells. Second, the approach is not to attack tumor-specific antigens but to remove an inhibitory pathway.
PD-1 plays a key role in inhibiting the effector function of antigen-specific CD8 T cells in chronic viral infections and cancer
In chronic viral infections and cancer, T lymphocytes are under persistent exposure to antigen stimulation. This is commonly associated with progressive deterioration of T cell effector function with constitutive coinhibitory receptor expression by T lymphocytes, a state called ‘exhaustion’. It usually manifests as a gradual loss of effector functions and cytokine production, as well as persistently increased expression of multiple inhibitory receptors [36,37,38]. T cell exhaustion was demonstrated in chronic viral infections such as human immunodeficiency virus (HIV), hepatitis C virus (HCV), and hepatitis B virus (HBV), and in cancer conditions [36, 38,39,40,41]. Exhausted T cells are characterized by deficits in proliferation and in the activation of effector functions (cytotoxicity and cytokine production) upon antigen stimulation [42]. Coinhibitory receptors are highly expressed on dysfunctional or exhausted T cells. The inhibitory ligands that regulate T cell function and induce T cell exhaustion/dysfunction in tissues usually exhibit increased expression on cancer cells and virus-infected cells within tissue microenvironments.
The immune checkpoint molecule PD-1 was originally identified from a T-cell line as a novel member of the immunoglobulin gene superfamily with an immunologic receptor tyrosine-based inhibitory motif (ITIM) [43]. Initially, PD-1 was demonstrated to be a receptor for cell death; however, the PD-1 pathway was later found to play a regulatory role in inhibiting T cell activation and restraining T cell function [12, 13]. Accumulating evidence indicates that the PD-1 pathway is critical in inhibiting viral antigen-specific CD8 T cells in chronic HIV [44], HCV [45], and HBV infections [25, 46]. Recent studies demonstrated that the interaction between PD-1 on T cells and its ligands plays an important role in inducing T cell exhaustion and dysfunction. Restoration of T cell function by PD-1 blockade supported the importance of this inhibitory pathway in animal models of viral infection [25, 41, 47, 48]. Moreover, it was shown that targeting PD-1 and other immune checkpoints is able to reverse this dysfunctional state and reinvigorate T cell activity in chronic viral infections and cancer [6, 24, 36, 38, 41, 49, 50].
Multiple inhibitory receptors are expressed by “exhausted” T cells in cancer and chronic viral infections
Whereas inhibitory receptors can be transiently expressed by effector T cells during the activation stage; persistent overexpression of inhibitory receptors is a hallmark of exhausted T cells [51,52,53,54]. So far, the molecular mechanisms by which inhibitory receptors regulate T cell exhaustion are still unclear. In addition to PD-1, exhausted T cells also express multiple inhibitory receptor molecules on their cell surface [42]. These inhibitory receptors include the lymphocyte activation gene 3 (LAG-3) protein, T cell immunoglobulin- and mucin-containing molecule-3 (Tim-3), CTLA4, and many other inhibitory receptors [49]. In fact, a core set of inhibitory receptors, including PD-1, LAG-3,Tim-3, and the T cell immunoglobulin and ITIM domain (TIGIT, also known as Vstm3 and WUCAM), is also expressed on tumor-infiltrating lymphocytes (TILs). Other combinations of inhibitory receptors, such as PD-1 and Tim-3 [55, 56] are also co-expressed in exhausted/dysfunctional T cells to regulate their function. Taken together, the accumulating results on these inhibitory receptors in co-regulation of T cell dysfunction suggest that these coinhibitory pathways may play different roles in T cell exhaustion.
Recent genomic studies exploring the transcriptional profile underlying T cell exhaustion revealed that exhausted T cells have a transcriptional profile with major alterations in the expression of inhibitory receptors, cytokine and chemokine receptors, signaling molecules, transcription factors, and genes involved in T cell metabolism [37, 57, 58]. Although considerable advances in mechanistic study have been made in the past few years, the molecular mechanisms of T cell dysfunction/exhaustion are still not clear. In addition, there still lacks a clear understanding of the intriguing molecular pathways involved in the reversal of T cell exhaustion/dysfunction. In fact, we have only just begun to understand the transcriptional coordination of T cell exhaustion. Moreover, accumulating studies have emphasized the pivotal importance of T cell metabolism in regulating T cell dysfunction/exhaustion [59,60,61]. This has prompted intense exploration into the targeting of other immune checkpoints or coinhibitory receptors besides PD-1 and CTLA4. Among them, LAG-3, Tim-3, and TIGIT are emerging immune checkpoints under preclinical and clinical development for cancer therapy.
LAG-3
Among the new immune checkpoints, LAG-3 was originally cloned in 1990 as a membrane protein upregulated on activated T lymphocytes, and natural killer (NK) cells [62]. LAG-3 gene has high homology with CD4 and structurally resembles the CD4 molecule. Similarly, LAG-3 binds to MHC class II with a higher affinity [63]. In addition to MHC class II, LSECtin, a DC-SIGN family molecule, was suggested to be another ligand for LAG-3 [64]. The most well known feature and function of LAG-3 are its role in the negative regulation of T cell response, and this makes it a potential target for immune modulation. LAG-3 is highly expressed on both activated natural regulatory T cells (nTreg) and induced FoxP3+ Treg (iTreg) cells [65]. Blockade of LAG-3 abolishes the suppressor function of Treg cells. Moreover, LAG-3 is crucial for Treg cell-mediated T cell homeostasis [66, 67]. All these results support a functional role for LAG-3 in Treg cell function. In cancer and chronic viral infections, expression of LAG-3 is increased in exhausted T cells [49]. PD-1 and LAG-3 are co-expressed on dysfunctional or exhausted virus-specific CD8+ T cells [68], and on both CD4+ and CD8+ tumor infiltrating lymphocytes (TILs) in animal models of cancer [69]. Blockade of LAG-3 can enhance anti-tumor T cell responses [70]. Co-blockade of the LAG-3 and PD-1 pathways is more effective for anti-tumor immunity than blocking either molecule alone [69, 71]. Therefore, in both chronic viral infections and cancer, PD-1 and LAG-3 signaling pathways functionally cooperate to inhibit T lymphocytes responses. The potential for LAG-3-driven immuno-modulatory responses are currently being explored for clinical cancer therapy.
Tim-3
Tim-3, another newly defined immune checkpoint, was first identified as a T cell surface molecule expressed selectively in interferon (IFN)-γ-producing T cells [72]. It is also expressed in innate immune cells (DCs, NK cells, and monocytes) and Treg cells [73]. Tim-3 blockade was shown to exacerbate experimental autoimmune encephalomyelitis (EAE) [72]. Studies with Tim-3 knockout mice and wild-type mice treated with Tim-3-blocking antibody demonstrated that Tim-3 signaling is required for tolerance induction and that Tim-3 blockade enhances the development of autoimmunity [74, 75]. Galectin-9, a C-type lectin, was first discovered as a Tim-3 ligand [76]. Triggering of Tim-3 by galectin-9 induced the death of Tim-3+ T cells and reduced EAE disease severity [76]. Most recently, CEACAM-1 was also identified as a novel cell surface ligand for Tim-3 [77]. CEACAM-1 co-immunoprecipitates with Tim-3, and it is co-expressed with Tim-3 on CD8+ TILs that exhibit the dysfunctional/exhausted phenotype. The regulatory function of Tim-3 is abrogated in the absence of CEACAM-1, suggesting a requirement of CEACAM-1/Tim-3 co-expression and interaction for optimal Tim-3 function [77].
The interleukin (IL)-27/NFIL3 axis was identified as a crucial regulator of effector function of T lymphocytes via induction of Tim-3 and the immunosuppressive cytokine IL-10 [78]. Tim-3's function in T cell exhaustion was recently examined in both chronic viral infections and cancer. The observation that Tim-3+ CD8+ T cells exhibit the dysfunctional/exhausted phenotype raised the question of whether PD-1 expression can be used as the sole hallmark for identifying dysfunctional/exhausted CD8+ T cells in chronic viral infections or cancer. In HIV infection, Tim-3 was found on dysfunctional/exhausted T cells that lacked PD-1 expression. Furthermore, Tim-3 was expressed in the most dysfunctional/exhausted population among CD8+PD-1+ T cells in several chronic viral (HCV and HBV) infections in humans and also in animal models [55, 79,80,81]. All these observations suggest that PD-1 and Tim-3 have non-redundant and synergistic functions in inhibiting effector T cell activity. In addition, studies on Tim-3 also indicate the presence of dysfunctional/exhausted CD8+ T cells in cancer. It was shown that populations of CD8+ TILs expressing both Tim-3 and PD-1display different functional phenotypes. Among these populations, Tim-3+PD-1+ double-positive TILs exhibit more dysfunctional or exhausted phenotypes than do Tim-3+ or PD-1+ single-positive TILs. In contrast, Tim-3-PD-1 double-negative TILs exhibit good effector function [56]. In support of these observations, co-blockade of the PD-1 and Tim-3 pathways was shown to be a more effective approach than blocking PD-1 alone for improving antitumor function and suppressing tumor progression in preclinical models of cancer. Taken together, the current data suggest that Tim-3 plays a crucial role in regulating antitumor T cell immunity [56, 82, 83].
TIGIT
TIGIT, a recently defined new immune checkpoint, was first identified as a novel CD28 family molecule [84,85,86,87]. TIGIT is an immunoglobulin (Ig) superfamily receptor that functions as a coinhibitory receptor, and is specifically expressed by immune cells [85,86,87]. TIGIT is expressed by activated T cells, and is also expressed on Treg cells, memory T cells, NK cells, and follicular T helper (Tfh) cells [84,85,86,87,88,89]. TIGIT binds two ligands, CD112 (PVRL2, nectin-2) and CD155 (PVR), and these ligands are expressed by T cells, APCs, and tumor cells [84,85,86, 90, 91]. Genome-wide association studies have linked TIGIT to multiple human autoimmune diseases including type 1 diabetes, multiple sclerosis, and rheumatoid arthritis [92, 93]. The function of TIGIT was therefore initially investigated in autoimmunity and tolerance. In addition to its protective role in autoimmune diseases, TIGIT was also explored in cancer and chronic viral infections. The TIGIT ligands, CD112 and CD155, are expressed in many tumor cells. In addition, the positive counterpart of this costimulatory pathway, CD226, promotes cytotoxicity and enhances antitumor responses [94, 95]. The TIGIT-deficient mice showed significantly delayed tumor progression in different tumor models, suggesting that TIGIT negatively regulates antitumor responses [96]. TIGIT is highly expressed on TILs in the tumor microenvironment across a broad range of tumors [96,97,98]. TIGIT+CD8+ TILs co-express PD-1, LAG-3, and Tim-3 and exhibit the most dysfunctional phenotype among CD8+ TILs in murine tumors [96]. TIGIT synergizes with PD-1 and also with Tim-3 in impairing anticancer immunity [96]. Therefore, co-blockade of either TIGIT plus PD-1 or TIGIT plus Tim-3 enhances anti-cancer immunity and induces tumor regression. Taken together, these results indicate that TIGIT synergizes with other inhibitory molecules to suppress effector T cell responses and promote T cell dysfunction.
Immune effector cells acquire inhibitory receptors in the tumor microenvironment
The ligands and inhibitory receptors that regulate T cell effector functions are mostly overexpressed on tumor-infiltrating immune cells or on tumor cells in the tumor microenvironment. Therefore, targeting these ligands and receptors is relatively specific to tumors compared to normal tissues. It is within these tumor microenvironments that immune effector cells acquire inhibitory receptors, resulting in T cell dysfunction. Soluble molecules include cytokines with immunosuppressive activity, such as IL-10, transforming growth factor (TGF)-β, and IL-27, which regulate immune responses to tumor cells and induce T cell dysfunction within the tumor microenvironment [99,100,101,102]. The IL-10 pathway has been intensively studied for its role in T cell dysfunction in chronic viral infections and cancer [99, 100]. IL‑10 promotes T cell exhaustion, and IL‑10 blockade reverses T cell dysfunction during chronic viral infections [99]. Co-blockade of both IL-10 and the PD1 reverses CD8+ T cell exhaustion and enhances viral clearance, which supports a role for IL-10 in T cell exhaustion [101]. Moreover, inhibition of TGFβ signalling in CD8+ T cells in vitro restores the dysfunction of exhausted T cells [103]. However, systemic blockage of TGFβ by treatment with its inhibitor or blocking antibody had only little benefit [104]. The type I IFNs (IFNα/β) are crucial in innate antiviral effects; however, recent reports demonstrated that type I IFNs signaling paradoxically facilitated viral persistence by enhancing immune suppression during chronic infection, and IFNα/β blockade reversed T cell exhaustion in chronic viral infection [105, 106], All these data suggest a possible role for IFNα/β in promoting exhaustion. Thus, chronic exposure to IFNα/β enhances T cell exhaustion/dysfunction during chronic infections. In recent studies, it has been demonstrated that the immunosuppressive cytokine, IL-27, is a potent inducer of Tim-3+ exhausted/dysfunctional T cells and a promoter of tumor growth in mice model [78]. Furthermore, IL-27 signaling directly controls expression of Tim-3 via induction of NFIL3, a transcription factor, which is critical for the development of exhausted/dysfunctional T cell phenotype [78]. Moreover, IL-27 is an inducer of the “coinhibitory” gene module in effector T cells. IL-27 induces inhibitory molecules including PD-1, Tim-3, LAG-3, TIGIT, and IL-10, which overlap with the mediators of T cell exhaustion, in chronic viral infections and cancer [101, 102]. From these observations, IL-27 signaling pathway may regulate the suppression program that drives the development of T cell exhaustion in cancer and chronic viral infections.
Besides inhibitory receptors on the cell surface, there are other soluble immune-inhibitory molecules within the tumor microenvironment. These soluble immune inhibitory molecules include certain metabolic enzymes, such as arginase produced by myeloid-derived suppressor cells (MDSCs), and indoleamine 2,3‑dioxygenase (IDO), which are expressed by both cancer cells and tumor-infiltrating myeloid cells [107,108,109,110]. Moreover, FOXP3+ CD4+ Treg cells also influence effector T cell function in the microenvironment within tumor. However, exactly how Treg cells affect the induction of T cell dysfunction has not been well defined. Besides FOXP3+CD4+ Treg cells, other immune cell types, such as NK cells, immunoregulatory APCs, MDSCs [111, 112], and CD8+ regulatory cells [113, 114], may affect tumor progression and directly or indirectly enhance T cell dysfunction.
CTLA4 and PD-1, the two immune checkpoint targets that have been extensively studied in clinical immuno-oncology, regulate anticancer T cell responses via different mechanisms and at different levels. This implies that anticancer immunity can be enhanced at multiple levels and by different mechanisms. It also implies that combination strategies for cancer immunotherapy can be wisely designed based on mechanisms and on results obtained from preclinical models. A better understanding of the specialized regulatory roles of these receptors and definition of the mechanisms of T cell dysfunction will provide more insights for rational design and development of cancer immunotherapy that target these receptors.
Conclusion
Recent studies demonstrated that immune checkpoint inhibitors are able to induce durable, long-lasting cancer control. These antibodies have moved immuno-oncology therapy into a new era and indicate that modulation of immune response is a crucial therapeutic strategy for cancer treatment. Although current immunotherapies targeted at the immune checkpoints, PD-1 and CTLA-4, exhibit enormous potential to control cancer, there are still some tumor types and many patients that remain largely refractory to these therapies. This has prompted intense investigation into the targeting of other immune checkpoints or coinhibitory receptors in order to increase the therapeutic repertoire. A diverse array of T cell co-receptors are now being explored for developing new potential targets for clinical cancer therapies. There are multiple additional immune checkpoints which represent new potential targets for cancer immunotherapy, and they are now under active development. These include antibodies targeting new immune checkpoints, particularly LAG-3, Tim-3, and TIGIT [73]. They also include agonist antibodies against activating receptors such as CD137, CD27, ICOS, GITR, B7-H3, and others. Rational combinations of immune checkpoint inhibitors with other immunotherapies are also tested in ongoing studies [19]. In addition, new biomarkers that help to select patients for particular types of immune checkpoint therapy are under intense investigations. A clearer understanding of the regulatory roles of these immune checkpoints and elucidation of the mechanisms of T cell dysfunction will shed insights on the development of new therapies for cancer treatment.
Abbreviations
- APC:
-
Antigen presenting cells
- CTLA-4:
-
Cytotoxic T lymphocytes antigen- 4
- DC:
-
Dendritic cell
- EAE:
-
Experimental autoimmune encephalomyelitis
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HIV:
-
Human immunodeficiency virus
- IFN:
-
Interferon
- LAG-3:
-
Lymphocyte activation gene 3 protein
- MDSC:
-
Myeloid-derived suppressor cells
- MHC:
-
Major histocompatibility complex
- NK cell:
-
Natural killer cell
- PD-1:
-
Programmed death-1
- TCR:
-
T-cell receptor
- TIGIT:
-
T cell immunoglobulin and ITIM domain
- TIL:
-
Tumor infiltrating lymphocyte
- Tim-3:
-
T cell immunoglobulin- and mucin-containing molecule-3
- Treg:
-
Regulatory T cells
References
Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N Engl J Med. 2016;375:143–53.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.
Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:1270–1.
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17.
Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–7.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54.
Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff Jr CL, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol. 2016;17:943–55.
Robert C, Soria JC, Eggermont AM. Drug of the year: programmed death-1 receptor/programmed death-1 ligand-1 receptor monoclonal antibodies. Eur J Cancer. 2013;49:2968–71.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–5.
Wing K, Yamaguchi T. Sakaguchi S Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. 2011;32:428–33.
Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–47.
Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, et al. T-cell tolerance or function is determined by combinatorial costimulatory signals. Embo J. 2006;25:2623–33.
Nishimura H, Nose M, Hiai H, Minato N. Honjo T Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51.
Kasagi S, Kawano S. Kumagai S PD-1 and autoimmunity. Crit Rev Immunol. 2011;31:265–95.
Song YW, Im CH, Park JH, Lee YJ, Lee EY, et al. T-cell immunoglobulin and mucin domain 3 genetic polymorphisms are associated with rheumatoid arthritis independent of a shared epitope status. Hum Immunol. 2011;72:652–5.
Wang M, Ji B, Wang J, Cheng X, Zhou Q, et al. Tim-3 polymorphism downregulates gene expression and is involved in the susceptibility to ankylosing spondylitis. DNA Cell Biol. 2014;33:723–8.
Zhang Q, Vignali DA. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity. 2016;44:1034–51.
Callahan MK, Postow MA, Wolchok JD. Targeting T Cell Co-receptors for Cancer Therapy. Immunity. 2016;44:1069–78.
Eggermont AM, Kroemer G, Zitvogel L. Immunotherapy and the concept of a clinical cure. Eur J Cancer. 2013;49:2965–7.
Pauken KE, Wherry EJ. SnapShot: T Cell Exhaustion. Cell. 2015;163:1038. e1031.
Attanasio J, Wherry EJ. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity. 2016;44:1052–68.
Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–84.
Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–76.
Tzeng HT, Tsai HF, Liao HJ, Lin YJ, Chen L, et al. PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PLoS ONE. 2012;7:e39179.
Sharpe AH, Abbas AK. T-cell costimulation--biology, therapeutic potential, and challenges. N Engl J Med. 2006;355:973–5.
Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–82.
Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143–55.
Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–94.
Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–10.
Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16:23–35.
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53.
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332:600–3.
Read S, Malmstrom V. Powrie F Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302.
Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–10.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–9.
Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, et al. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–44.
Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35:51–60.
Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–13.
Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. 1998;187:1383–93.
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99.
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo J. 1992;11:3887–95.
Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4.
Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, et al. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81:9249–58.
Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–25.
Maier H, Isogawa M, Freeman GJ, Chisari FV. PD-1:PD-L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J Immunol. 2007;178:2714–20.
Velu V, Titanji K, Zhu B, Husain S, Pladevega A, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–10.
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37.
Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3--potential mechanisms of action. Nat Rev Immuno. 2015;15:45–56.
Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14:1212–8.
Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–45.
Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol. 2012;188:2957–65.
Araki K, Youngblood B, Ahmed R. Programmed cell death 1-directed immunotherapy for enhancing T-cell function. Cold Spring Harb Symp Quant Biol. 2013;78:239–47.
Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010;107:14733–8.
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–94.
Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity. 2014;40:289–302.
Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–84.
Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454.
Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. 2013;14:1173–82.
Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8(+) T cells during chronic infection. Immunity. 2014;41:802–14.
Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171:1393–405.
Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol. 1995;25:2718–21.
Xu F, Liu J, Liu D, Liu B, Wang M, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014;74:3418–28.
Huang CT, Workman CJ, Flies D, Pan X, Marson AL, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–13.
Workman CJ, Vignali DA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223). J Immunol. 2005;174:688–95.
Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, et al. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004;172:5450–5.
Richter K, Agnellini P, Oxenius A. On the role of the inhibitory receptor LAG-3 in acute and chronic LCMV infection. Int Immunol. 2010;22:13–23.
Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–27.
Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. 2007;117:3383–92.
Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107:7875–80.
Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415:536–41.
Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004.
Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immuno. 2003;4:1102–10.
Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol. 2003;4:1093–101.
Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6:1245–52.
Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517:386–90.
Zhu C, Sakuishi K, Xiao S, Sun Z, Zaghouani S, et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat Commun. 2015;6:6072.
Ju Y, Hou N, Zhang XN, Zhao D, Liu Y, et al. Blockade of Tim-3 pathway ameliorates interferon-gamma production from hepatic CD8+ T cells in a mouse model of hepatitis B virus infection. Cell Mol Immunol. 2009;6:35–43.
McMahan RH, Golden-Mason L, Nishimura MI, McMahon BJ, Kemper M, et al. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J Clin Invest. 2010;120:4546–57.
Nebbia G, Peppa D, Schurich A, Khanna P, Singh HD, et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE. 2012;7:e47648.
Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, et al. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71:3540–51.
Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117:4501–10.
Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. 2011;41:902–15.
Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S. 2009;106:17858–63.
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10:48–57.
Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol. 2009;39:695–703.
Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. 2011;186:1338–42.
Joller N, Lozano E, Burkett PR, Patel B, Xiao S, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40:569–81.
Casado JG, Pawelec G, Morgado S, Sanchez-Correa B, Delgado E, et al. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol Immunother. 2009;58:1517–26.
Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–65.
Hafler JP, Maier LM, Cooper JD, Plagnol V, Hinks A, et al. CD226 Gly307Ser association with multiple autoimmune diseases. Genes Immun. 2009;10:5–10.
Maiti AK, Kim-Howard X, Viswanathan P, Guillen L, Qian X, et al. Non-synonymous variant (Gly307Ser) in CD226 is associated with susceptibility to multiple autoimmune diseases. Rheumatology (Oxford). 2010;49:1239–44.
Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, et al. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J Exp Me. 2008;205:2965–73.
Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, et al. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J Exp Med. 2008;205:2959–64.
Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125:4053–62.
Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. 2015;125:2046–58.
Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26:923–37.
Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, et al. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–9.
Brooks DG, Walsh KB, Elsaesser H, Oldstone MB. IL-10 directly suppresses CD4 but not CD8 T cell effector and memory responses following acute viral infection. Proc Natl Acad Sci U S A. 2010;107:3018–23.
Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell. 2016;166:1500–11. e1509.
Wu C, Pot C, Apetoh L, Thalhamer T, Zhu B, et al. Metallothioneins negatively regulate IL-27-induced type 1 regulatory T-cell differentiation. Proc Natl Acad Sci U S A. 2013;110:7802–7.
Tinoco R, Alcalde V, Yang Y, Sauer K, Zuniga EI. Cell-intrinsic transforming growth factor-beta signaling mediates virus-specific CD8+ T cell deletion and viral persistence in vivo. Immunity. 2009;31:145–57.
Garidou L, Heydari S, Gossa S, McGavern DB. Therapeutic blockade of transforming growth factor beta fails to promote clearance of a persistent viral infection. J Virol. 2012;86:7060–71.
Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–11.
Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–7.
Mellor AL, Keskin DB, Johnson T, Chandler P, Munn DH. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J Immunol. 2002;168:3771–6.
Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. 2002;101:151–5.
Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–54.
Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91.
Ng CT, Snell LM, Brooks DG, Oldstone MB. Networking at the level of host immunity: immune cell interactions during persistent viral infections. Cell Host Microbe. 2013;13:652–64.
Goh C, Narayanan S, Hahn YS. Myeloid-derived suppressor cells: the dark knight or the joker in viral infections? Immunol Rev. 2013;255:210–21.
Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2012;481:394–8.
Holderried TA, Lang PA, Kim HJ, Cantor H. Genetic disruption of CD8+ Treg activity enhances the immune response to viral infection. Proc Natl Acad Sci U S A. 2013;110:21089–94.
Acknowledgements
The authors thank Dr. I-Tsu Chyuan and Dr. Chien-Sheng Wu for critical reviews and discussions of this article.
Funding
This work was supported by grants from the National Science Council, Taiwan (NSC 101-2321-B-002-008, and 104-2314-B-281 -002 -; MOST 105-2320-B-002-034 -, and 105-2320-B-038-065 -).
Availability of data and materials
Data and materials related to this work are available upon request.
Authors’ contributions
HFT and PNH wrote the review article. Both authors read and approved the manuscript. PNH obtained funding.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All authors approve the manuscript for publication.
Ethics approval and consent to participate
This review article does not involve ethics approval and consent to participate.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tsai, HF., Hsu, PN. Cancer immunotherapy by targeting immune checkpoints: mechanism of T cell dysfunction in cancer immunity and new therapeutic targets. J Biomed Sci 24, 35 (2017). https://doi.org/10.1186/s12929-017-0341-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-017-0341-0