
Abstract
Background
Avian influenza virus (AIV), infectious bronchitis virus (IBV), and Newcastle disease virus (NDV) are important avian pathogens that can cause enormous economic loss on the poultry industry. Different respiratory etiological agents may induce similar clinical signs that make differential diagnosis difficult. Importantly, AIV brings about severe threat to human public health. Therefore, a novel method that can distinguish these viruses quickly and simultaneously is urgently needed.
Results
In this study, an oligonucleotide microarray system was developed. AIV, including H5, H7, and H9 subtypes; NDV; and IBV were simultaneously detected and differentiated on a microarray. Three probes specific for AIV, NDV, and IBV, as well as three other probes for differentiating H5, H7, and H9 of AIV, were first designed and jet-printed to predetermined locations of initiator-integrated poly(dimethylsiloxane) for the synchronous detection of the six pathogens. The marked multiplex reverse transcription polymerase chain reaction (PCR) products were hybridized with the specific probes, and the results of hybridization were read directly with the naked eyes. No cross-reaction was observed with 10 other subtypes of AIV and infectious bursal disease virus, indicating that the oligonucleotide microarray assay was highly specific. The sensitivity of the method was at least 100 times higher than that of the conventional PCR, and the detection limit of NDV, AIV, H5, H7, and H9 can reach 0.1 EID50 (50% egg infective dose), except that of IBV, which was 1 EID50 per reaction. In the validation of 93 field samples, AIV, IBV, and NDV were detected in 53 (56.99%) samples by oligonucleotide microarray and virus isolation and in 50 (53.76%) samples by conventional PCR.
Conclusions
We have successfully developed an approach to differentiate AIV, NDV, IBV, H5, H7, and H9 subtypes of AIV using oligonucleotide microarray. The microarray is an accurate, high-throughput, and relatively simple method for the rapid detection of avian respiratory viral diseases. It can be used for the epidemiological surveillance and diagnosis of AIV, IBV, and NDV.
Similar content being viewed by others


Background
At present, the demand for broiler meat products is increasing worldwide; however, avian viruses cause respiratory infections and lead to mortality in poultry flocks, which impose negative influence on chicken products [1]. The avian influenza virus (AIV), Newcastle disease virus (NDV), and infectious bronchitis virus (IBV) can cause enormous economic loss in the poultry industry worldwide. AIV belongs to the Orthomyxoviridae family, Influenza A virus genus. The AIV genome is composed of eight segments of negative-sense single-stranded RNA and classified into different subtypes according to two surface antigens: hemagglutinin (HA) and neuraminidase (NA); heretofore, there are 18 known HA and 11 known NA subtypes [2]. Among all known HA subtypes, the highly pathogenic avian influenza viruses have been restricted to H5 and H7, which not only cause considerable economic losses in the poultry industry [3] but also seriously threaten human health [4]. Other subtypes cause a milder respiratory disease, and they are designated as low pathogenicity avian influenza (LPAI) viruses. Although H9 is an LPAI, it is the dominant circulating subtype, showing a high prevalence [5]. NDV belongs to the Paramyxoviridae family, Avulavirus genus, and is an RNA-containing virus. It appears to be a sporadic epizootic disease despite vaccination programs [6]. In industrial poultry farms, birds infected with NDV must be immediately sacrificed due to the threat of infection dissemination across countries. IBV belongs to the family Coronaviridae and the genus Coronavirus. It infects chickens of all ages and causes lesions in respiratory and urogenital organs [7]. Although the IBV vaccine plays a vital role in controlling IB, IBV outbreaks frequently occur and are still considered a global epidemic [8].
These viruses, in association with bacterial agents or independently, can lead to diseases [9, 10]. In addition, the clinical symptoms caused by single- or multiple-viruses are similar, which makes it difficult for veterinarians to distinguish these viruses in the field. Therefore, sensitive and rapid detection techniques that can distinguish these respiratory viral infections are needed for the surveillance of the emergence of new viruses, outbreak management, and disease control.
Currently, virus isolation combined with hemagglutination inhibition and NA inhibition tests is considered to be the conventional gold standard; however, it is time consuming (it needs four to 6 days), has low sample throughput, and is relatively insensitive [11]. Polymerase chain reaction (PCR) is widely used to identify the aforementioned viruses via molecular diagnostic assays, and reverse transcription PCR (RT-PCR) is now becoming accepted as a new gold standard in many studies because it shows superior sensitivity [12, 13]. The real-time PCR method, which can quantitate samples, is also used; however, it requires expensive equipment [14]. Moreover, most of the aforementioned methods are used for testing only one agent in a specimen [15]. Multiplex RT-PCR assays that can involve simultaneous amplification of more than one infectious agent are also available, the advantages of the multiplex RT-PCR combine the sensitivity and speed of PCR and eliminate the need for testing clinical samples for each virus separately [16, 17]. Many studies have also been performed using multiplex real-time RT-PCR to differentiate single or mixed avian virus infection, such as NDV, AIV, and IBV [18,19,20,21]. In recent years, microarray technology has also been developed [22, 23].
The correct differentiation between viral infections is very important for accurately monitoring and effectively controlling respiratory avian diseases. Herein, we developed a rapid, concurrent, and high-throughput oligonucleotide microarray approach for the detection of avian respiratory pathogens, such as AIV, NDV, and IBV single- and mixed-virus infections, including the H5, H7, and H9 subtypes of AIV.
Methods
Plasmids, viruses and clinical specimens
The standard plasmids (pMD18-T-AIV-M, pMD18-T-H5, pMD18-T-H7, pMD18-T-H9, pMD18-T-NDV-F, pMD18-T-IBV-N) were obtained by our laboratory. The virus strains used in this study, including AIV, NDV, IBV, and IBDV, were obtained from the Key Animal Virology Laboratories of the Ministry of Agriculture of China and were preserved at − 80 °C (Table 1).
A total of 93 samples, including oropharyngeal and cloacal swabs, were collected from the live poultry markets of the Jiangsu Province, China. The oropharyngeal swab or cloacal swab was dipped respectively in 2 mL phosphate-buffered saline to release the fecal or tracheal materials from the swabs. Each specimen was separated into two halves: one half by extracting viral RNA was used to perform microarray and reverse transcription of cDNA for traditional PCR, while the other half was used for virus isolation. Virus isolation of these clinical samples were isolated using specific-pathogen-free embryonated hen’s eggs (Tianbang Biotechnology, China), then the type and subtype were identified by reverse transcription and conventional PCR assay, respectively [24, 25].
Primers and probes
Sequences of virus nucleotides were downloaded from the National Center for Biotechnology Information GenBank (See Additional file 2: Table S1). The common probes and primers targeting AIV, NDV, and IBV were designed from the conserved sequences of AIV-M gene, NDV-F gene, and IBV-N gene, respectively. The probes and primers of AIV H5, H7, and H9 subtypes were based on conserved sequences of HA gene. The probes of H7 and H9 subtypes and the primers of H5, H7, and H9 subtypes have already been designed [24].
The selected nucleotide sequences were aligned by using Lasergene (DNASTAR Inc., USA), and these primers were designed using Primer Express 3.0 (Applied Biosystems, USA). All the designs for forward primers were labeled with biotin on the 5′ end. The primers and probes are listed in Table 2.
RNA extraction and cDNA synthesis
The viral RNAs of AIV, NDV, and IBV strains were extracted by using the RNeasy mini kit (Qiagen Inc., CA) according to the manufacturer’s instructions. The concentration and purity of the extracted total RNA were determined by measuring the absorbance ratio at a wavelength of 260 nm over 280 nm using a NanoDrop 2000c spectrophotometer (Thermo Scientific, USA). The final extracted pellets were stored at − 80 °C.
First-strand cDNA was synthesized using AMV reverse transcriptase (TaKaRa Biotechnology, China). Briefly, the following reagents were added and mixed: 4 μL of 5× reverse transcriptase buffer, 2 μL of dNTP mixture (10 mmol/L), 1 μL of random primer (50 mmol/L), 2 μL of AMV reverse transcriptase, 0.5 μL of RNAase inhibitor (40 U/μL), 5 μL of RNA, and 5.5 μL of RNAase-free water. The reaction mixture was sequentially incubated at 42 °C for 60 min and then at 72 °C for 15 min. The cDNA was subsequently stored at − 20 °C.
Microarray printing
Microarray was prepared in a 100,000 grade clean room. Probes were diluted with printing buffer (0.3 M phosphate buffer, 0.2% glycerin, 0.01% Triton X-100, and 1.5% mannitol) for further printing. Each dilution of probes was printed on initiator-integrated poly(dimethylsiloxane) (iPDMS), a novel solid supporting material. The oligonucleotide microarray was completed using a contact printer SmartArrayer 48 (CapitalBio, China) with approximately 0.6 nL of printing solution for each sample. Each well has positive control with biotin and negative control with printing buffer.
Multiplex RT-PCR
The multiplex RT-PCR was conducted in a 20.0 μL reaction system with a PCR machine (Eppendorf, Germany). Multiplex RT-PCR primers were adapted as previously described for AIV, NDV, and IBV (Table 2). Multiplex RT-PCR was performed using HiScript® II One Step qRT-PCR Probe Kit (Vazyme, USA). The reaction mixture contained 2.0 μL mixed primers (IBV-F, IBV-R, NDV-F, NDV-R, M-F, M-R, H5-F, H5-R, H7-F, H7-R, H9-F, H9-R), 10.0 μL of 2× One-Step Q Probe Mix, 1.0 μL One-Step Q Probe Enzyme Mix, 6.0 μL of RNase-free water, and 1.0 μL of extracted RNA (total: 20.0 μL). The multiplex RT-PCR program was as follows: a reverse transcription step at 50 °C for 5 min, initial denaturation at 95 °C for 2 min, and finally 40 amplification cycles of 95 °C for 10 s and 54 °C for 10 s.
Hybridization reaction
The multiplex RT-PCR product was diluted 1:4 with 2× saline sodium citrate (SSC) + 0.1% sodium dodecyl sulfate (SDS) (wash A). The diluting product was denatured in boiling water bath for 5 min and immediately cooled in an ice bath for 2 min. The microarray was preheated at 37 °C for 5 min. Then, 60 μL of the diluting product was added to the microarray chamber, incubated at 47 °C, stirred at 200 rpm for 20 min, and washed twice with 0.5× SSC + 0.2% SDS (wash B, the preheating maintenance temperature of wash B must be equal to 47 °C). Streptavidin–horseradish peroxidase (streptavidin-HRP) was diluted 1:500 with wash A. A dilute solution of streptavidin-HRP should be preheated to 47 °C, and 60 μL should be added to the microarray chamber. The oligonucleotide microarray was incubated on a shaker plate at 47 °C for 20 min at 200 rpm, washed three times with wash B, and then washed twice with 0.05 M sodium citrate buffer (wash C, room temperature). A total of 100 μL tetramethylbenzidine chromogenic reagent was added onto the microarray and was allowed to stand for 3 min to read the results with the naked eyes.
Conventional PCR and real-time PCR
A total of 20 μL of conventional PCR mixture included 10.0 μL of 2× Taq Master Mix (Vazyme, USA), 0.2 μM of each pair of primers (Table 2), 1.0 μL of template and ddH2O. The standard plasmids (pMD18-T-AIV-M, pMD18-T-H5, pMD18-T-H7, pMD18-T-H9, pMD18-T-NDV-F, pMD18-T-IBV-N) served as a positive control, while ddH2O played the role of the negative control. The thermocycling parameters were as follows: 95 °C for 5 min, 40 cycles of 95 °C for 30 s, 54 °C for 30 s, and 72 °C for 1 min, followed by 72 °C for 10 min at the end of the reaction. PCR product was separated on a 1.0% agarose gel. The assay was repeated at least 2 times and two technical replicates were run for each assay.
The real-time PCR was performed in a 20.0 μL reaction system with a LightCycler 96 real-time PCR system (Roche, Switzerland). The 20.0 μL reaction mixture comprised 10.0 μL of SYBR Premix Ex Taq II (TaKaRa Biotechnology, China), 2.0 μL of each pair of primers (Table 2), 2.0 μL of template, and 6.0 μL of ddH2O. The real-time PCR program was as follows: predenaturation at 95 °C for 30 s and 40 amplification cycles of 95 °C for 5 s and 54 °C for 30 s. The positive and negative controls (pMD18-T-AIV-M, pMD18-T-H5, pMD18-T-H7, pMD18-T-H9, pMD18-T-NDV-F, pMD18-T-IBV-N and ddH2O) of real-time PCR were set. Fluorescent signals were obtained once per cycle upon the completion of the extension step. Samples exhibiting a cycle threshold (Ct) value of less than 35 were considered positive. The assay was repeated at least 2 times and two technical replicates were run for each assay.
Specificity and sensitivity of the oligonucleotide microarray
The primers and probes were evaluated by using the BLAST tool, and the specificity of the oligonucleotide microarray was evaluated by cross-reactivity with other subtypes (H1, H2, H3, H4, H6, H8, H10, H11, H12, H13 AIVs) of influenza A virus and IBDV. The sensitivity of the method was evaluated using tenfold serial dilution of virus quantified by 50% egg infective dose (EID50). The final concentration of virus RNA was between 105 and 10− 2 EID50 per reaction mixture. Each dilution of virus and negative control was tested by conventional PCR, oligonucleotide microarray, and real-time PCR.
Reproducibility of the oligonucleotide microarray assay
To evaluate the reproducibility of the oligonucleotide microarray assay, equal amounts of five virus strains (H5, H7, H9, IBV, and NDV) were mixed, and the titer of each virus was 106 EID50 per reaction. The mixture was tested in three independent reactions over separate days.
Application of the oligonucleotide microarray assay
Before detecting the specimens, the co-infection models, which included various arrangements, were designed and tested to determine the detection efficiency of the oligonucleotide microarray.
The 93 clinical oropharyngeal and swabs were assayed to evaluate the feasibility of the methods for detection of AIV; IBV; NDV; and H5, H7, and H9 subtypes.
Results
Multiplex RT-PCR
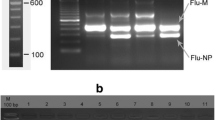
The study developed a multiplex RT-PCR with six pairs of primers for the fragment of NDV-F, IBV-N, AIV-M, AIV-H5, AIV-H7, and AIV-H9 prior to the microarray test. The PCR product was 163 bp (AIV-M), 210 bp (AIV-H5), 105 bp (AIV-H7), 157 bp (AIV-H9), 107 bp (NDV), and 175 bp (IBV) (See Additional file 1: Figure S1).
Oligonucleotide microarray assay
A single virus or various combinations of the viruses (AIV, NDV, and IBV) were tested using the oligonucleotide microarray assay following the multiplex RT-PCR. All viruses were detected and typed expressly, and no cross-reaction with other probes was found (Fig. 1). In this study, the results were clearly recognizable with the naked eyes, and no accessional imaging equipment was needed. This finding indicated that the simultaneous detection, differentiation, and typing of AIV, NDV, and IBV can be inexpensively and easily achieved using the oligonucleotide microarray.

Detection and typing of NDV, IBV, or AIV using oligonucleotide microarrays. a Microarray map. Each dot indicates the spotted position of each probe. P: Positive control; N: Negative control; 1: AIV-H5; 2: AIV-H7; 3: AIV-H9; 4: AIV-M; 5: IBV-N; 6: NDV-F. b Detection and typing results shown on the microarrays. B1: H5 AIV; B2: H7 AIV; B3: H9AIV; B4: IBV; B5: NDV; B6: H5 AIV + NDV; B7: H7 AIV + NDV; B8: H7 AIV + IBV + NDV; B9: H9 AIV + IBV; B10: H9 AIV + NDV; B11: H5 AIV + H7 AIV; B12: H5 AIV + H9 AIV + NDV; B13: H5 AIV + H7 AIV + H9 AIV; B14: H5 AIV + H7 AIV + H9 AIV + IBV + NDV; B15: Negative control
Specificity and sensitivity of the oligonucleotide microarray assay
The oligonucleotide array had good specificity, and no cross-reactivity with any of the other avian respiratory viruses was found. The oligonucleotide microarray assay was examined by testing H1–H13 (except H5, H7, and H9) AIVs and IBDV. The influenza viruses could react with the M probe only (Fig. 2).

Specificity of oligonucleotide microarrays. Rapid detection of the other avian respiratory viruses and H1–H13 AIVs using oligonucleotide microarrays. 1: Positive control; 2: H1 AIV; 3: H2 AIV; 4: H3 AIV; 5: H4 AIV; 6: H6 AIV; 7: H8 AIV; 8: H10 AIV; 9: H11 AIV; 10: H12 AIV; 11: H13 AIV; 12: IBDV; 13: Negative control
The detection limit comparison test among conventional PCR assay, oligonucleotide microarray, and the real-time PCR was performed. The results showed that the conventional PCR assay could be seen on agarose gel when infectious virus titer was 102 EID50 per reaction, except for NDV, which showed positive results only when the virus titer was more than 103 EID50 per reaction (Table 3). The detection limit of real-time PCR was 0.1 EID50 per reaction with the H5 and NDV, and others were approximately 1 EID50 per reaction (Table 3). Moreover, the sensitivity of the microarray relative to the detectable infectious virus titer was 0.1 EID50 per reaction with the NDV, M, H5, H7, and H9. The detection limit of IBV could reach 1 EID50 (Table 3). This finding showed that the oligonucleotide microarray sensitivity was at least 100 times higher than that of the conventional PCR in this study, consistent with or even slightly better than real-time PCR.
Reproducibility of the oligonucleotide microarray assay
To evaluate the reproducibility of the oligonucleotide microarray assay, one mixture of five virus strains was tested in three independent reactions over separate days. Upon visual inspection of three independent reactions, all pathogens could be detected. The results showed that oligonucleotide microarray assay has high reliability and reproducibility.
Co-infection models
The co-infection results proved that this method could detect mixed infection accurately, whether it was AIV, IBV, and NDV triplex infection or between each of the multiplex infections. In the mixture proportion of each component at the same level and in the gap, the method could accurately detect all the viruses in the co-infection system (Table 4).
Clinical specimen detection, diagnostic sensitivity, and diagnostic specificity
A total of 93 field samples were analyzed using the oligonucleotide microarray, conventional PCR, and virus isolation assay. The results showed that the respective positive detection rates with conventional PCR, oligonucleotide microarray, and the virus isolation were 30.11, 33.33, and 33.33% for AIV and 23.66% for NDV (Table 5). For AIV, a total of 31 positive samples among 93 clinical samples were detected with oligonucleotide microarray and virus isolation, while 28 positive samples were detected with conventional PCR. For NDV, 22 positive samples were detected, and no obvious difference was found for the identification of these methods. None of the field samples was IBV positive. Oligonucleotide microarray and virus isolation showed higher detection rates of H5, H7, and H9 than conventional PCR. However, these methods had the same detection rate for NDV and IBV. Moreover, using the microarray method, two samples were found to be co-infected with H7 and H9 AIVs, and no other co-infected samples were observed in the study. The results of AIV, NDV, and IBV detection in the specimens by different methods were compared and showed that microarray detection could be applied for laboratory surveillance and diagnosis of the pathogens in clinical fields.
Virus isolation was used as a standard method in our studies. The diagnostic sensitivity and specificity were both 100% for the oligonucleotide microarray, whereas those for the conventional PCR were 94.33 and 100%, respectively (Table 6).
Discussion
AIV, NDV, and IBV are highly contagious pathogens with high incidence in poultry [26, 27]. They pose a considerable threat to the poultry industry [28]. For diagnosing avian viral infection, many methods have been developed. For example, virus isolation, RT-PCR, real-time RT-PCR, fluorescent antibody test, and enzyme-linked immunosorbent assay are the currently used methods for the laboratory diagnosis of avian viruses. However, each of these methods has its advantages and limitations. The disadvantages of these methods include the following: time consuming, nonspecific, expensive, or labor-intensive [12, 29, 30]. To establish clinical diagnosis, accurate and fast multiplex detection of avian respiratory viruses is very important. In recent years, multiplex RT-PCR and microarray technology are resoundingly applied to detect AIV and its subtypes and diagnose multiple infections such as combination of NDV and AIV [31].
The diagnosis of virus types based on microarray technology can improve the quality and shorten the analysis duration in molecular diagnosis of infectious diseases. Moreover, it can satisfy the needs of simultaneous detection of multiple viruses and screening large numbers of pathology samples [32, 33]. Microarray assays for avian diseases have been previously reported. For disease diagnosis, there are oligonucleotide microarrays that can screen not only AIV and NDV but also IBV and IBDV [23]. A DNA suspension array-based assay for avian respiratory viruses, which can identify AIV, NDV, IBV, and infectious laryngotracheitis virus, has also been reported [34]. Electronic microarray assays have been used to simultaneously detect all AIV H and N subtypes and pathotyping of NDV [22].
In the present study, a microarray technology-based iPDMS with “absolute” zero background was developed, which can simplify data analysis and reduce nonspecific interactions [35, 36]. This is the first time to use this material to detect the nucleotides of avian diseases. In addition, 48 samples can be detected at once, and the results can be read visually. The microarray can simultaneously detect AIV, NDV, and IBV and accurately distinguish AIV haemagglutinin subtypes H5, H7, and H9, whereas virus isolation in embryonated eggs and conventional PCR can only detect one agent in a sample. The duration of this assay without time required for the viral RNA extraction is 2–3 h, and 48 specimens can be simultaneously assayed. We evaluated the capability of the developed assay by comparing its sensitivity with conventional PCR and real-time PCR. The virus titer was 1 EID50 per reaction, which can be detected by microarray. The result clearly shows that the limits of detection reached 1 EID50 per reaction for NDV and 0.1 EID50 per reaction for M, H5, H7, H9, and IBV, which was at least 100 times more sensitive than that of conventional PCR. Furthermore, the detection limit of oligonucleotide microarray is the same as real-time PCR for H5 and NDV and more sensitive than real-time PCR for IBV and H7, H9, and M of AIV. The detection results of the co-infection model also proved that the method established in this study can be used for the accurate detection of mixed infection, which, to a certain degree, also shows the viability of this method. Moreover, 93 avian field samples were used to test the effectiveness and reliability of the microarray, and the experimental results were consistent with the virus isolation. Furthermore, the co-infection situation can be detected in clinical practice by the microarray. The diagnostic sensitivity and specificity of the oligonucleotide microarray were both 100%.
In summary, the microarray assay provides an alternative high-throughput molecular diagnostic platform for susceptive and specific detection of several major viruses generally seen as vital causes of viral respiratory diseases in poultry. The microarray assay proves to be time-saving in that the entire experiment process, including PCR, took only 2.5 h. Compared with other methods, the microarray assay is more efficient because it can detect three viruses and identify HA subtypes of AIV. It is also convenient because the results of the experiments can be determined with the naked eyes, disregarding the need for expensive experimental apparatus and complicated analysis.
Conclusions
The oligonucleotide microarray with high sensitivity and specificity was successfully developed for the rapid detection of AIV, NDV, and IBV. More importantly, it can simultaneously identify H5, H7, and H9 subtypes of AIV. This assay will be a useful tool in the control and management of AIV, NDV, and IBV.
Availability of data and materials
The dataset analyzed during the current study is available from the corresponding author on reasonable request.
References
Sakai K, Yada K, Sakabe G, Tani O, Miyaji K, Nakamura M, Takehara K. Serological and virological studies of Newcastle disease and avian influenza in slaughter-age ostriches (Struthio camelus) in Japan. J Vet Med Sci. 2006;68(5):491–4.
Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, et al. New world bats harbor diverse influenza a viruses. PLoS Pathog. 2013;9(10):e1003657.
Dhingra MS, Artois J, Dellicour S, Lemey P, Dauphin G, Von Dobschuetz S, Van Boeckel TP, Castellan DM, Morzaria S, Gilbert M. Geographical and historical patterns in the emergences of novel highly pathogenic avian influenza (HPAI) H5 and H7 viruses in poultry. Front Vet Sci. 2018;5:84.
Li X, Zhang Z, Yu A, Ho SY, Carr MJ, Zheng W, Zhang Y, Zhu C, Lei F, Shi W. Global and local persistence of influenza a(H5N1) virus. Emerg Infect Dis. 2014;20(8):1287–95.
Ma MJ, Zhao T, Chen SH, Xia X, Yang XX, Wang GL, Fang LQ, Ma GY, Wu MN, Qian YH, et al. Avian influenza a virus infection among Workers at Live Poultry Markets, China, 2013-2016. Emerg Infect Dis. 2018;24(7):1246–56.
Mariappan AK, Munusamy P, Kumar D, Latheef SK, Singh SD, Singh R, Dhama K. Pathological and molecular investigation of velogenic viscerotropic Newcastle disease outbreak in a vaccinated chicken flocks. Virusdisease. 2018;29(2):180–91.
Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet Res. 2007;38(2):281–97.
Zhou H, Zhang M, Tian X, Shao H, Qian K, Ye J, Qin A. Identification of a novel recombinant virulent avian infectious bronchitis virus. Vet Microbiol. 2017;199:120–7.
Malik YS, Patnayak DP, Goyal SM. Detection of three avian respiratory viruses by single-tube multiplex reverse transcription-polymerase chain reaction assay. J Vet Diagn Invest. 2004;16(3):244–8.
Ali A, Reynolds DL. A multiplex reverse transcription-polymerase chain reaction assay for Newcastle disease virus and avian pneumovirus (Colorado strain). Avian Dis. 2000;44(4):938–43.
Ziegler T, Hall H, Sanchez-Fauquier A, Gamble WC, Cox NJ. Type- and subtype-specific detection of influenza viruses in clinical specimens by rapid culture assay. J Clin Microbiol. 1995;33(2):318–21.
Hodinka RL. Point: is the era of viral culture over in the clinical microbiology laboratory? J Clin Microbiol. 2013;51(1):2–4.
Letant SE, Ortiz JI, Bentley Tammero LF, Birch JM, Derlet RW, Cohen S, Manning D, McBride MT. Multiplexed reverse transcriptase PCR assay for identification of viral respiratory pathogens at the point of care. J Clin Microbiol. 2007;45(11):3498–505.
Arya M, Shergill IS, Williamson M, Gommersall L, Arya N, Patel HR. Basic principles of real-time quantitative PCR. Expert Rev Mol Diagn. 2005;5(2):209–19.
Stone B, Burrows J, Schepetiuk S, Higgins G, Hampson A, Shaw R, Kok T. Rapid detection and simultaneous subtype differentiation of influenza a viruses by real time PCR. J Virol Methods. 2004;117(2):103–12.
Munch M, Nielsen LP, Handberg KJ, Jorgensen PH. Detection and subtyping (H5 and H7) of avian type a influenza virus by reverse transcription-PCR and PCR-ELISA. Arch Virol. 2001;146(1):87–97.
He J, Bose ME, Beck ET, Fan J, Tiwari S, Metallo J, Jurgens LA, Kehl SC, Ledeboer N, Kumar S, et al. Rapid multiplex reverse transcription-PCR typing of influenza a and B virus, and subtyping of influenza a virus into H1, 2, 3, 5, 7, 9, N1 (human), N1 (animal), N2, and N7, including typing of novel swine origin influenza a (H1N1) virus, during the 2009 outbreak in Milwaukee, Wisconsin. J Clin Microbiol. 2009;47(9):2772–8.
Acevedo AM, Perera CL, Vega A, Rios L, Coronado L, Relova D, Frias MT, Ganges L, Nunez JI, Perez LJ. A duplex SYBR green I-based real-time RT-PCR assay for the simultaneous detection and differentiation of Massachusetts and non-Massachusetts serotypes of infectious bronchitis virus. Mol Cell Probes. 2013;27(5–6):184–92.
Callison SA, Hilt DA, Boynton TO, Sample BF, Robison R, Swayne DE, Jackwood MW: Development and evaluation of a real-time Taqman RT-PCR assay for the detection of infectious bronchitis virus from infected chickens. J Virol Methods 2006;138(1–2):60–65.
Farkas T, Szekely E, Belak S, Kiss I. Real-time PCR-based pathotyping of Newcastle disease virus by use of TaqMan minor groove binder probes. J Clin Microbiol. 2009;47(7):2114–23.
Huber I, Campe H, Sebah D, Hartberger C, Konrad R, Bayer M, Busch U, Sing A. A multiplex one-step real-time RT-PCR assay for influenza surveillance. Euro Surveill. 2011;16(7).
Lung O, Beeston A, Ohene-Adjei S, Pasick J, Hodko D, Hughes KB, Furukawa-Stoffer T, Fisher M, Deregt D. Electronic microarray assays for avian influenza and Newcastle disease virus. J Virol Methods. 2012;185(2):244–53.
Sultankulova KT, Kozhabergenov NS, Strochkov VM, Burashev YD, Shorayeva KA, Chervyakova OV, Rametov NM, Sandybayev NT, Sansyzbay AR, Orynbayev MB. New oligonucleotide microarray for rapid diagnosis of avian viral diseases. Virol J. 2017;14(1):69.
Liu J, Yao L, Zhai F, Chen Y, Lei J, Bi Z, Hu J, Xiao Q, Song S, Yan L, et al. Development and application of a triplex real-time PCR assay for the simultaneous detection of avian influenza virus subtype H5, H7 and H9. J Virol Methods. 2018;252:49–56.
Motitschke A, Ottiger HP, Jungback C. Evaluation of the sensitivity of PCR methods for the detection of extraneous agents and comparison with in vivo testing. Biologicals. 2010;38(3):389–92.
Roussan DA, Khawaldeh GY, Shaheen IA. Infectious bronchitis virus in Jordanian chickens: seroprevalence and detection. Can Vet J. 2009;50(1):77–80.
Nguyen TT, Kwon HJ, Kim IH, Hong SM, Seong WJ, Jang JW, Kim JH. Multiplex nested RT-PCR for detecting avian influenza virus, infectious bronchitis virus and Newcastle disease virus. J Virol Methods. 2013;188(1–2):41–6.
Neumann G, Chen H, Gao GF, Shu Y, Kawaoka Y. H5N1 influenza viruses: outbreaks and biological properties. Cell Res. 2010;20(1):51–61.
Renaud C, Crowley J, Jerome KR, Kuypers J. Comparison of FilmArray respiratory panel and laboratory-developed real-time reverse transcription-polymerase chain reaction assays for respiratory virus detection. Diagn Microbiol Infect Dis. 2012;74(4):379–83.
Kuypers J, Campbell AP, Cent A, Corey L, Boeckh M. Comparison of conventional and molecular detection of respiratory viruses in hematopoietic cell transplant recipients. Transpl Infect Dis. 2009;11(4):298–303.
Wang LC, Pan CH, Severinghaus LL, Liu LY, Chen CT, Pu CE, Huang D, Lir JT, Chin SC, Cheng MC, et al. Simultaneous detection and differentiation of Newcastle disease and avian influenza viruses using oligonucleotide microarrays. Vet Microbiol. 2008;127(3–4):217–26.
Sultankulova KT, Chervyakova OV, Kozhabergenov NS, Shorayeva KA, Strochkov VM, Orynbayev MB, Sandybayev NT, Sansyzbay AR, Vasin AV. Comparative evaluation of effectiveness of IAVchip DNA microarray in influenza a diagnosis. TheScientificWorldJournal. 2014;2014:620580.
Liu LY, Ye HY, Chen TH, Chen TC. Development of a microarray for simultaneous detection and differentiation of different tospoviruses that are serologically related to tomato spotted wilt virus. Virol J. 2017;14(1):1.
Laamiri N, Fallgren P, Zohari S, Ben Ali J, Ghram A, Leijon M, Hmila I. Accurate detection of avian respiratory viruses by use of multiplex PCR-based Luminex suspension microarray assay. J Clin Microbiol. 2016;54(11):2716–25.
Ma H, Wu Y, Yang X, Liu X, He J, Fu L, Wang J, Xu H, Shi Y, Zhong R. Integrated poly(dimethysiloxane) with an intrinsic nonfouling property approaching "absolute" zero background in immunoassays. Anal Chem. 2010;82(15):6338–42.
Huang M, Ma Q, Liu X, Li B, Ma H. Initiator integrated poly(dimethysiloxane)-based microarray as a tool for revealing the relationship between nonspecific interactions and irreproducibility. Anal Chem. 2015;87(14):7085–91.
Acknowledgments
Not applicable.
Funding
The design of the oligonucleotide microarray method was supported by the National Key Research and Development Program of China (2016YFD0501600) and Key Program of Science and Technology Planning of Guangdong Province (2017B020202010). The sampling swabs of chicken was supported by the Natural Science Foundation of Jiangsu Province (BK20181321, BK20161452) and Forestry Science and Technology Innovation and Promotion Project of Jiangsu Province (LYKJ[2018]22). Writing of the report was supported by the China Agriculture Research System (CARS-40-K13) and analysis and interpretation of data was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
LPY and QX careful conceived and designed the study. QX, LY and JHH performed the experiments and analyzed the results. ZWB, YQC, AF, HL and LY collected the clinical samples. QX and LPY wrote the manuscript. LPY, YY and JYZ analyzed the data and revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. PCR products of F (NDV), N (IBV), M (AIV), H9 (AIV), H7 (AIV) and H5 (AIV) gene fragments. (DOCX 235 kb)
Additional file 2:
Table S1. Accession numbers of the GenBank reference sequences used to design the primer. (DOCX 27 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xiao, Q., Yan, L., Yao, L. et al. Development of oligonucleotide microarray for accurate and simultaneous detection of avian respiratory viral diseases. BMC Vet Res 15, 253 (2019). https://doi.org/10.1186/s12917-019-1985-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-019-1985-7