
Abstract
Background
As the epidemiology of human Q Fever generally reflects the spread of Coxiella burnetii in ruminant livestock, molecular characterization of strains is essential to prevent human outbreaks. In this study we report the genetic diversity of C. burnetii in central Italy accomplished by MST and MLVA-6 on biological samples from 20 goat, sheep and cow farms.
Results
Five MST and ten MLVA profiles emerged from the analysis establishing a part of C. burnetii strain world atlas. In particular, ST32 occurred on 12 farms (60%), prevalently in goat specimens, while ST12 (25%) was detected on 4 sheep and 1 goat samples. ST8 and a variant of this genotype were described on 2 different sheep farms, whereas ST55 was observed on a goat farm. Five complete MLVA profiles different from any other published genotypes were described in this study in addition to 15 MLVA incomplete panels. Despite this, polymorphic markers Ms23, Ms24 and Ms33 enabled the identification of samples sharing the same MST profile.
Conclusions
Integration of such data in international databases can be of further help in the attempt of building a global phylogeny and epidemiology of Q fever in animals, with a “One Health” perspective.
Similar content being viewed by others


Background
Coxiella burnetii is the causative agent of Query (Q) fever in humans, a zoonotic disease present throughout the world, except in New Zealand [1], and coxiellosis in animals [2]. It is an obligate intracellular bacterium, replicating in eukaryotic cells that shows a biphasic developmental cycle: the large cell variant (LCV) corresponding to the intracellular replicative form and the small cell variant (SCV) which represents the host cell free stable form that is highly resistant to different environmental stresses [3]. The SCVs may persist in the environment for years [1, 3]. C. burnetii is found in urine, faeces and milk of infected animals, although transmission to humans is most frequently due to the inhalation of aerosolized bacteria that are spread in the environment by infected animals after delivery or abortion. Amniotic fluid and placenta contain the highest concentration of bacteria [4]. In recent years, an increasing number of animals have been reported to shed the bacterium, including pets, reptiles, ticks, rodents and birds [5,6,7,8], however, the main reservoirs of C. burnetii are cattle, sheep, and goats [9, 10]. Because Q fever is a zoonosis, the epidemiology of human infections generally reflects the circulation of the bacterium in ruminant livestock and serological investigation. Prophylaxis and molecular characterization of the strains circulating is therefore essential in order to prevent human outbreaks. Several genotyping methods have been described thus far. Until 2005, these techniques were based on plasmid typing, restriction fragment length polymorphism followed by pulsed-field gel electrophoresis (RFLP-PFGE) analysis, and sequence analysis of individual genes (e.g. 16S). Nevertheless, some of these methods exhibited limitations on inter- and intra-laboratory reproducibility and poor discrimination power that hindered their widespread use [11]. In 2005, typing via sequence analysis of multi-spacer regions (MST) was introduced by Glazunova et al. [12], who identified 10 highly variable intergenic spacers allowing the unambiguous characterization of the agent. MST genotyping has high levels of discrimination and helps to trace the spread of C. burnetii from one region to another and to define phylogenetic relationships [13, 14]. Otherwise, multi-locus Variable Number Tandem Repeat (VNTR) analysis (MLVA) was first established by Svraka et al. [15], and then improved by Arricau-Bouvery et al. [16]. Since their rapid evolution VNTR are extremely polymorphic, therefore MLVA usually provide a better discriminatory power, which is often suitable for epidemiological purposes [17]. MST and MLVA are both PCR-based techniques, and they have the potential to be used directly on non-cultured samples, avoiding the culture step of the pathogen that requires biosafety level 3 and long-time analyses. In this study, we report the genetic diversity of C. burnetii by MST and MLVA with the aim of describing the strains circulating in central Italy, taking into account the knowledge acquired after the Q Fever outbreaks in the Netherlands [18,19,20].
Methods
Biological samples and DNA extraction
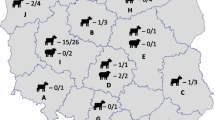
During the period 2012–2015 20 farms were positive for C. burnetii by Real Time PCR [21], with slight modifications [14]. Farms were named G1-G10 for goat, S1-S9 for sheep, and C1 for cow (Table 2). Brain, spleen, lung and liver were sampled from aborted goat (N. 10) and sheep (N. 9) fetuses and vaginal swabs were collected from the relative parturient animals. Placenta was collected only in two sampling sessions, these samples referred to a goat (G2) and sheep (S9). A milk sample was collected from a serological positive cow (C1). Only one DNA sample per farm was used for genotyping purposes; the selection was driven by the lowest Ct values obtained by Real Time PCR. Details of the samples per geographic origin, specimen, year of collection and Ct value are reported in Table 1 and Fig. 1.

Geographical localization of the farms in central Italy. Farm ID was designated according to animal species host: G = goat; S = sheep; C = cattle
DNA from fetal organs was isolated using the Maxwell 16 Tissue DNA Purification Kit (Promega) following the manufacturer’s instructions. Cells from milk were recovered by centrifuging 50 ml at 2000 g for 10 min; the pellet was resuspended with 300 μl of nuclease free water and then DNA was isolated using the Maxwell 16 Cell DNA Purification Kit (Promega) following the manufacturer’s instructions.
MST and MLVA
Multispacer Sequence Typing of C. burnetii DNA was performed on 20 specimens as previously described by Glazunova et al. [12] with some modifications [14]. Raw sequence data were assembled using SeqMan Pro (DNASTAR Lasergene 10 Core Suite). The coded alleles were compared with the sequences in the reference database available on the website http://ifr48.timone.univ-mrs.fr/mst/coxiella_burnetii/groups.html. MLVA was performed on the same samples for the 6 loci panel [16] with some modifications described by Tilburg et al. [22]. Furthermore, in order to obtain complete panels, new reverse primers were designed for the refractive markers Ms24 (5’-ACAAGCTATTTACTCCCTTTCTGC-3′) and Ms34 (5’-GCGTTAGTGTGCTTATCTCTTG-3′) by the Primer Express v3.0.1 software (Applied Biosystems), while for Ms23 and Ms33 primers from the website http://mlva.u-psud.fr/MLVAnet/spip.php?rubrique50 were selected. Three more DNA samples for which MST profile was previously reported [14], were included in the VNTR analysis for a total of 23 different strains. The amplification products were diluted 1:10 and 1:100 in distilled water and 1.2 μl of every single dilution was added to reaction mixture containing 10.5 μl formamide and 0.3 μl of Gene Scan 500 size standard marker (Applied Biosystems). PCR products were denatured for 30 s at 95 °C, cooled on ice for 2 min and then run on AB3130XL capillary sequencer with POP7 polymer. VNTR fragments were finally sized via GeneMapper software v.4.0 (Applied Biosystems). DNA from the Nine Mile strain (RSA 493) was used as a reference. According to the online database support site (http://mlva.u-psud.fr/mlvav4/genotyping/index.php), the MLVA profile of the Nine Mile strain is 9 27 4 6 9 5 for markers Ms23, Ms24, Ms27, Ms28, Ms33 and Ms34, respectively. For each marker the repeats number was determined inferring the sizes of the sample fragments with those obtained using the reference strain, run in the same time. Ten samples were sequenced to confirm the repeats number and the presence of Insertion Sequences (IS).
Results
The MST analysis from the 10 goat, 9 sheep and 1 cattle PCR-positive farms revealed two dominant previously described Sequence Types: ST32, reported in 12 farms (60%), and ST12, reported in 5 farms (25%). In addition, ST8 and ST55 were reported in the farms S5 (5%) and G1 (5%) respectively. Finally, a variant of the ST8 (proposed ST62) previously described only in this geographic area [14], was described in farm S1 (5%). This new allelic combination is 5 4 2 5 1 5 3 2 4 4 for the spacers Cox2, Cox5, Cox18, Cox20, Cox22, Cox37, Cox51, Cox56, Cox57, and Cox61 respectively. A complete panel was accomplished for all samples through MST analysis (Table 2, Fig. 2), while incomplete results were obtained for MLVA (Table 2). Although only partial panels were gained for the samples G1, G3-G6, G8-G10, S1-S5, S9 and C1, the high resolution power of MLVA allowed differentiation of strains sharing the same MST profile such as the case of the sample from farm G3, with ST32 like eight other farms, but showing a unique allele (12 tandem repeats) in the Ms24 marker. The case of strains from goat farms G7 and G8, located in the same geographic area and having the same ST32, is analogous. The variation in the number of tandem repeats in the locus Ms24 allowed the differentiation of the two genotypes showing 11 and 10 tandem repeats respectively, as well as the strains from farms S9 and G10 (11 and 12 tandem repeats, respectively), while showing the same ST12. Moreover, the polymorphism of the marker MS33 allows the discrimination of the strain G8 (9 tandem repeats) from G2 and G9 (5 tandem repeats + IS1111), all of these sharing the same ST32. Ten different MLVA genotypes emerged from the analysis (Table 2). Three samples are omitted from the MLVA description because of the limited amount of DNA available. The five complete MLVA profiles compared with those deposited in the public MLVA database (http://mlva.u-psud.fr/mlvav4/genotyping/index.php) did not match with any of the published genotypes. Sequence analysis of the samples G2, G7, and G9 confirmed the presence of the IS1111.

Genetic diversity. Sequence types are expressed accordingly to MST database. ST8+ refers to a variant of the ST8 as previously described (Di Domenico et al. [14])
Discussion
Discrimination of bacterial strains based on the analysis of their genetic content has become widely used due to its high resolution. Genetic variability can explain most of the phenotypic heterogeneity within bacterial population, such as host specificity, pathogenicity, antibiotic resistance, and virulence [23]. Currently, two main discriminant methods are commonly used for Coxiella burnetii genotyping: MST and MLVA [4]. MST has succeeded in establishing reliable correlations of STs with geographic distribution, clinical manifestations, and epidemiology of strains. Despite some STs being found worldwide, many others are restricted to specific areas. As a result, it has also been qualified as a “geotyping method” [4]. For example, to date ST17 has been isolated only from French Guiana, where it causes severe forms of the disease [24]. Acute Q Fever patients from French Guiana demonstrate significantly higher phase I IgG titers, whereas phase II antibodies are generally revealed at this stage of the disease [4]. Of note, pneumonia is observed in 83% of the patients representing the genotype with the highest prevalence of community-acquired pneumonia in the world [4, 25]. Analogously, ST33 is distributed in different small areas of Europe including Germany, where it was first described, and the Netherlands where it spread via France, causing the largest Q Fever outbreak ever described [26, 27]. Strains belonging to the ST23 group were reported in ticks, birds, ruminants and humans only in a restricted area between Eastern Europe (Czech Republic and Slovakia) and Asian countries (Russia, Kazakhstan, Mongolia and Uzbekistan), (http://ifr48.timone.univ-mrs.fr/mst/coxiella_burnetii/strains.html). Another interesting case is that of ST21. Despite two isolates from France and one from the United States, it is mainly reported in Nova Scotia [12]. On the other hand, some sequence types are distributed worldwide, such as ST16.
In Italy, sequence types already described are ST16, ST18 and ST29 (http://ifr48.timone.univmrs.fr/mst/coxiella_burnetii/strains.html), a novel sequence type similar to ST20, an additional sequence type related to ST8 and ST55 discovered in bovine milk, goat fetus and goat milk, respectively [14]. ST12 and ST32 are closely related on the basis of phylogenetic analysis [13, 14] and largely distributed along the area considered in the present study. Indeed, these two genotypes recur in 85% of the specimens (17/20) in cow, sheep and goat. These findings confirmed the spread of these genotypes in Tuscany as previously reported [28]. Interestingly, ST12 has been detected in clinical human samples from France, Switzerland and Senegal (http://ifr48.timone.univmrs.fr/mst/coxiella_burnetii/strains.html), whereas in animals it has been only found in Italy [28]. This result suggests that goat and sheep could represent an important source of human Q fever in this country. Although the oral exposure is still controversial [9], the transmission of C. burnetii to humans through inhalation of contaminated aerosol is widely recognized, especially for certain at-risk categories, such as farmers, veterinarians, or people living close to or exposed to infected flocks [29,30,31,32]. Conversely, the zoonotic origin of ST32 and the transmission to human have been already assessed. Indeed, as reported in the database http://ifr48.timone.univ-mrs.fr/mst/coxiella_burnetii/strains.html, it was identified in a goat placenta in Austria, and detected in human heart valve and aortic biopsy in Germany and France, respectively. Our study confirms ST32 detection in sheep specimens and it firstly describes the occurrence of this zoonotic genotype in cows. Three additional MST profiles have been reported in the present study: ST8, ST55 and a novel ST closely related to the ST8 previously identified in the same area [14]. Notably, ST8 was responsible for two human chronic Q Fever cases in Portugal [33].
The Q Fever outbreak in the Netherlands pushed toward the molecular investigation by both MST and MLVA of C. burnetii strains in different countries, not only in Europe. As VNTRs are important source of genetic polymorphisms for strain typing due to their rapid evolution, MLVA approach is particularly useful for epidemiological purposes. Unfortunately, PCR amplification is not always successful for all markers, so that partial genotypes are frequently obtained causing underestimation of the genotypic diversity [33,34,35,36]. Moreover, insertions or deletions may impair the estimation of the number of tandem repeats [37]. In particular, Ms23 and Ms33 both harbour a recognition site for the IS1111 insertion sequence upstream the repeat units and therefore may constitute preferred targets for insertions [37].
In the present study, five complete MLVA profiles were gained, all of which were different from the previously reported genotypes, including those recently found in Italy [35], while 15 panels were incomplete. Despite this, MLVA enabled 10 genotypes to be identified, instead of the 5 obtained by MST. Moreover, by means of sequencing we detected the presence of the IS1111 within the repetitive region of the Ms33 marker in three different goat samples sharing the same sequence type (ST32).
Conclusions
Our study based on MST and MLVA-6, established a part of C. burnetii strain atlas in central Italy. Integration of such data with international databases can be of further help in the attempt of building a global phylogeny and epidemiology of Q fever in animals, with a “One Health” perspective.
Abbreviations
- LCV:
-
Large cell variant
- MLVA:
-
Multi Locus VNTR Analysis
- MST:
-
Multispacer Sequence Typing
- RFLP-PFGE:
-
Restriction fragment length polymorphism followed by pulsed-field gel electrophoresis
- SCV:
-
Small cell variant
- ST:
-
Sequence Type
- VNTR:
-
Variable Number Tandem Repeat
References
Raoult D, Marrie TJ, Mege JL. Natural history and pathophysiology of Q fever. Lancet Infect Dis. 2005;5:219–26.
Pearson T, Hornstra HM, Hilsabeck R, Gates LT, Olivas SM, Birdsell DM, Hall CM, German S, Cook JM, Seymour ML, Priestley RA, Kondas AV, Clark Friedman CL, Price EP, Schupp JM, Liu CM, Price LB, Massung RF, Kersh GJ, Keim P. High prevalence and two dominant host-specific genotypes of Coxiella burnetii in U.S. milk. BMC Microbiol. 2014;14:41.
Tissot-Dupont H, Raoult D. Q Fever. Infect Dis Clin N Am. 2008;22:505–14.
Eldin C, Mélenotte C, Mediannikov O, Ghigo E, Million M, Edouard S, Mege JL, Maurin M, Raoult D. From Q fever to Coxiella burnetii infection: a paradigm change. Clin Microbiol Rev. 2017;30:115–90.
Pascucci I, Di Domenico M, Dall’Acqua F, Sozio G, Cammà C. Detection of Lyme disease and Q fever agents in wild rodents in central Italy. Vector-Borne Zoonotic Dis. 2015;15(7):404–11.
D’amato F, Million M, Edouard S, Delerce J, Robert C, Marrie T, Raoult D. Draft genome sequence of Coxiella burnetii Dog Utad, a strain isolated from a dog-related outbreak of Q fever. New Microbes New Infect. 2014;2:136–7.
Langley JM, Marrie TJ, Covert A, Waag DM, Williams JC. Poker players’ pneumonia. An urban outbreak of Q fever following exposure to a parturient cat. N Engl J Med. 1988;319(6):354–6.
Socolovschi C, Reynaud P, Kernif T, Raoult D, Parola P. Rickettsiae of spotted fever group, Borrelia valaisiana, and Coxiella burnetii in ticks on passerine birds and mammals from the Camargue in the south of France. Ticks Tick-borne Dis. 2012;3:354–9.
Scientific Opinion on Q fever. EFSA panel on Animal Health and Welfare (AHAW) EFSA panel on Biological Hazards (BIOHAZ) (Chapter 4 on Food Safety). EFSA J. 2010;8(5):1595.
Agerholm J. Coxiella burnetii associated reproductive disorders in domestic animals-a critical review. Acta Vet Scand. 2013;55:13.
Massung RF, Cutler SJ, Frangoulidis D. Molecular typing of Coxiella burnetii (Q fever). Adv Exp Med Biol. 2012;984:381–96.
Glazunova O, Roux V, Freylikman O, Sekeyova Z, Fournous G, Tyczka J, Tokarevich N, Kovacova E, Marrie TJ, Raoult D. Coxiella burnetii genotyping. Emerg Infect Dis. 2005;11(8):1211–7.
Hornstra HM, Priestley RA, Georgia SM, Kachur S, Birdsell DN, Hilsabeck R, Gates LT, Samuel JE, Heinzen RA, Kersh GJ, Keim P, Massung RF, Pearson T. Rapid typing of Coxiella burnetii. PLoS One. 2011;6(11):e26201.
Di Domenico M, Curini V, De Massis F, Di Provvido A, Scacchia M, Camma C. Coxiella burnetii in Central Italy: novel genotypes are circulating in cattle and goats. Vector-Borne Zoonotic Dis. 2014;14(10):710–5.
Svraka S, Toman R, Skultety L, Slaba K, Homan WL. Establishment of a genotyping scheme for Coxiella burnetii. Microbiol Lett. 2006;254(2):268–74.
Arricau-Bouvery N, Hauck Y, Bejaoui A, Frangoulidis D, Bodier C, Souriau A, Meyer H, Neubauer H, Rodolakis A, Vergnaud G. Molecular characterization of Coxiella burnetii isolates by infrequent restriction site-PCR and MLVA typing. BMC Microbiol. 2006;6:38.
van Belkum A. Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA). FEMS Immunol Med Microbiol. 2007;49:22–7.
Roest HIJ, Ruuls RC, Tilburg JJHC, Nabuurs-Franssen MH, Klaassen CHW, Vellema P, van den Brom R, Dercksen D, Wouda W, Spierenburg MAH, van der Spek AN, Buijs R, de Boer AG, Willemsen PTJ, van Zijderveld FG. Molecular epidemiology of Coxiella burnetii from ruminants in Q fever outbreak, the Netherlands. Emerg Infect Dis. 2011;17(4):668–75.
Roest HIJ, Tilburg JJHC, van der Hoek W, Vellema P. The Q fever epidemic in the Netherlands: history, onset, response and reflection. Epidemiol Infect. 2011;139:1):1–12.
Tilburg JJHC, Rossen JWA, van Hannen EJ, Melchers WJG, Hermans MHA, van de Bovenkamp J, Roest HJIJ, de Bruin A, Nabuurs-Franssen MH, Horrevorts AM, Klaassen CHW. Genotypic diversity of Coxiella burnetii in the 2007-2010 Q fever outbreak episodes in the Netherlands. J Clin Microbiol. 2012;50(3):1056–8.
Panning Panning M, Kilwinski J, Greiner-Fischer S, Peters M, Kramme S, Frangoulidis D, Meyer H, Henning K, Drosten C. High throughput detection of Coxiella burnetii by real-time PCR with internal control system and automated DNA preparation. BMC Microbiol. 2008;8:77.
Tilburg JJHC, Roest HJIJ, Nabuurs-Franssen MH, Horrevorts AM, Klaassen CHW. Genotyping reveals the presence of a predominant genotype of Coxiella burnetii in consumer milk products. J Clin Microbiol. 2012;50(6):2156–8.
Li W, Raoult D, Fournier PF. Bacterial strain typing in the genomic era. FEMS Microbiol. 2009;33:892–916.
Mahamat A, Edouard S, Demar M, Abboud P, Patrice JY, La Scola B, Okandze A, Djossou F, Raoult D. Unique clone of Coxiella burnetii causing severe Q fever, French Guiana. Emerg Infect Dis. 2013;19(7):1102–4.
D’Amato F, Eldin C, Georgiades K, Edouard S, Delerce J, Labas N, Raoult D. Loss of TSS1 in hypervirulent Coxiella burnetii 175, the causative agent of Q fever in French Guiana. Comp Immunol Microbiol Infect Dis. 2015;41:35–41.
Tilburg JJHC, Roest HJIJ, Buffet S, Nabuurs-Franssen MH, Horrevorts AM, Raoult D, Klaassen CHW. Epidemic genotype of Coxiella burnetii among goats, sheep, and humans in the Netherlands. Emerg Infect Dis. 2012;18(5):887–8.
D’Amato F, Rouli L, Edouard S, Tyczka J, Million M, Robert C, Nguyen TT, Raoult D. The genome of Coxiella burnetii Z3055, a clone linked to the Netherlands Q fever outbreaks, provides evidence for the role of drift in the emergence of epidemic clones. Comp ImmunolMicrobiol Infect Dis. 2014;37:281–8.
Galiero A, Fratini F, Cammà C, Di Domenico M, Curini V, Baronti I, Turchi B, Cerri D. Occurrence of Coxiella burnetii in goat and ewe unpasteurized cheeses: screening and genotyping. Int J Food Microbiol. 2016;237:47–54.
Manfredi Selvaggi T, Rezza G, Scagnelli M, Rigoli R, Rassu M, De Lalla F, Pellizzer GP, Tramarin A, Bettini C, Zampieri L, Belloni M, Pozza ED, Marangon S, Marchioretto N, Togni G, Giacobbo M, Todescato A, Binkin N. Investigation of a Q-fever outbreak in northern Italy. Eur J Epidemiol. 1996;12(4):403–8.
Starnini G, Caccamo F, Farchi F, Babudieri S, Brunetti B, Rezza G. An outbreak of Q fever in a prison in Italy. Epidemiol Infect. 2005;133:377–80.
Van den Brom R, van Engelen E, Roest HI, van der Hoek W, Vellema P. Coxiella burnetii infections in sheep or goats: an opinionated review. Vet Microbiol. 2015;181(1–2):119–29.
Brooke RJ, Mutters NT, Péter O, Kretzschmar ME, Teunis PF. Exposure to low doses of Coxiella burnetii caused high illness attack rates: insights from combining human challenge and outbreak. Epidemic. 2015;11:1–6.
Santos AS, Tilburg JJHC, Botelho A, Barahona MJ, Núncio MS, Nabuurs-Franssen MH, Klaassen CHW. Genotypic diversity of clinical Coxiella burnetii isolates from Portugal based on MST and MLVA typing. Int J Med Microbiol. 2012;302:253–6.
Račić I, Spičić S, Galov A, Duvnjak S, Zdelar-Tuk M, Vujnović A, Habrun B, Cvetnić Z. Identification of Coxiella burnetii genotypes in Croatia using multi-locus VNTR analysis. Vet Microbiol. 2014;173(3–4):340–7.
Ceglie L, Guerrini E, Rampazzo E, Barberio A, Tilburg JJHC, Hagen F, Lucchese L, Zuliani F, Marangon S, Natale A. Molecular characterization by MLVA of Coxiella burnetii strains infecting dairy cows and goats of North-Eastern Italy. Microbes Infect. 2015;17(11–12):776–81.
Sulyok KM, Kreizinger Z, Hornstra HM, Pearson T, Szigeti A, Dán Á, Balla E, Keim PS, Gyuranecz M. Genotyping of Coxiella burnetii from domestic ruminants and human in Hungary: indication of various genotypes. BMC Vet Res. 2014;10:107.
Sidi-Boumedine K, Duquesne V, Prigent M, Yang E, Joulié A, Thiéry R, Rousset E. Impact of IS1111 insertion on the MLVA genotyping of Coxiella burnetii. Microbes Infect. 2015;17(11–12):789–94.
Acknowledgements
The authors wish to thank Luigi Sorbara and Fiorentino Stravino for outstanding technical assistance.
Funding
Funding was provided by the Italian Ministry of Health. The funding organization played no role in the design of study, choice of specimens, review and interpretation of data, or preparation or approval of manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
All authors have made substantial contributions to conception and design of the study and have been involved in revising the manuscript critically for important intellectual content. Samples collection and acquisition of data for diagnosis purposes have been accomplished by LDG, AF, AC and AB. MDD, VC, CC, VDL and MM have made substantial contributions to acquisition and analysis of genotyping data. MDD have made significant contributions to interpretation of data and together with all other authors have been involved in drafting the manuscript. All authors have given final approval of the version to be published. Finally, all authors agree all aspects of the work ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Non-experimental research has been conducted on animals. Diagnostic investigations and laboratory analysis have been performed on samples that are submitted to the laboratory by both private and state veterinarians. The specimens used in this study are field samples originating from naturally aborted foetuses and sent to our laboratory for official diagnosis.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Di Domenico, M., Curini, V., Di Lollo, V. et al. Genetic diversity of Coxiella burnetii in domestic ruminants in central Italy. BMC Vet Res 14, 171 (2018). https://doi.org/10.1186/s12917-018-1499-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-018-1499-8