
Abstract
Background
Porcine circovirus type 1 (PCV1) was discovered in 1974 as a contaminant of a porcine kidney (PK-15) cell line and was generally accepted to be nonpathogenic. But recently it was shown to cause lesions in experimentally infected pig fetuses. Serological evidence and genetic studies suggested that PCV1 was widespread in domestic pigs. Thus, the molecular epidemiology and genetic variation of PCV1 are still necessary to understand.
Results
Here 247 tissue samples were collected from piglets in Guangxi Province, China and performed whole-genome sequencing of the PCV1 genome. Thirteen PCV1 strains were sequenced from the samples. Similarity analysis showed that there were 97.8% to 99.6% nucleotide similarity to each other and 97.1% to 99.8% nucleotide similarity to the 40 reference strains. Besides, based on sequence analysis, we found one putative recombinant virus named GXdx84 strain contained the open-reading frame 1 (ORF1) of PCV1 and the ORF2 of PCV2d-2, which was consistent with the results of phylogenetic analysis that compared PCV1 and PCV2 strains. Variation analysis of the amino acids of the capsid protein revealed that the GXyl224 strain, which encoded 235 amino acids, had two amino acids more than other strains. This is the first study to report that a cap gene mutation resulted in lengthening of in the gene sequence.
Conclusions
These data contribute to the understanding of PCV1 evolution and molecular epidemiology that will facilitate programs for its control and prevention.
Similar content being viewed by others


Background
Porcine circovirus 1 (PCV1) is a small, non-enveloped, circular single-stranded DNA virus with a genome length of 1.7kb. It is a member of the family Circoviridae and genus Circovirus [1, 2]. PCV has two genotypes: PCV1 and PCV2. PCV1 was first identified as a contaminant in a pig kidney cell culture (PK-15) [3, 4]. PCV2 is the causative agent of porcine circovirus-associated disease (PCVAD) in swine, and result in substantial economic losses for the pig industry in the world [5].
The genome organization of PCV1 and PCV2 are highly similar. The cis-acting and trans-acting replication factors of both viruses are interchangeable for DNA replication [6,7,8]. The overall DNA sequence similarity within the PCV1 or PCV2 isolations is greater than 90%, while the similarity between PCV1 and PCV2 isolations is 68% to 76%. There are two major open-reading frames, ORF1 and ORF2, which diverge from the Ori expressing four proteins [9]. ORF1 (rep gene) is transcribed in a clockwise direction and encodes two viral replication-associated proteins Rep and Rep'. These two proteins are the main factors for initiation of viral DNA replication. ORF2 (cap gene) is transcribed in an anti-clockwise direction, and encodes the immunogenic capsid protein which builds the capsid of the virus [10, 11]. In addition to the replication ORF1 and the capsid protein ORF2, another fragment recognized to be involved in modulation of virulence was encoded by ORF3. PCV1 and PCV2 utilize similar initiation and termination signals at comparable locations within the viral genome. However, they differ from each other with respect to specific RNA expression level as well as splice-junction selection is unique to each virus. Previous research shows that thirteen RNAs reported as PCV1, while ten RNAs to be PCV2 during virus replication in PK-15 cells [12, 13]. Besides, the cap of PCV2 encodes the viral capsid protein (Cap) and ORF3-RNA which encodes the apoptosis-associated protein. While the functions of the respective protein are the same, ORF3 of PCV1 is 612nt in length, twice the size of PCV2 [13].
Although PCV1 DNA has been isolated from lymph nodes of a piglet with a wasting condition, it is generally accepted that PCV1 is nonpathogenic but widespread in pigs and porcine cell line PK-15 [14, 15]. However, PCV1 can produce pathology in the lungs of porcine fetuses in foetal life [16]. Clinical data shows that PCV1 infection is common in pigs and that pigs can produce antibodies against PCV1 [6, 17, 18].
The main objective of this study is to analyze the prevalence of PCV1 and the evolutionary patterns as well as the relationships among PCV1 genomes isolated from Guangxi Province of China and compare them with data on PCV1 and recombination of PCV1 and PCV2 published worldwide.
Methods
Clinical samples
A total of 247 spleen and lymph node samples were collected from piglets (All piglets were euthanized by an anesthetic overdose with the pentobarbital before collected the samples) in Guangxi Province, China, in 2015. All the pigs displayed signs of progressive weight loss, inguinal lymph node edema and hemorrhage, pulmonary edema, and other lesions. Clinical tissues were homogenized for DNA extraction and stored at -80 °C.
DNA isolation and polymerase chain reaction (PCR)
Viral DNA was extracted using a TIANamp Genomic DNA kit (TIANGEN, Beijing, China). Two pair of specific PCR primers named PCV1-F and PCV1-R, PCV2-F and PCV2-R were designed according to published PCV1 and PCV2 sequences to amplify the complete genome (Table 1). The PCR assays were performed in a 25μL reaction mixture consisting of 3ng tissue-isolated DNA templates, containing final concentrations of 1.25mM MgCl2, 2.5μL 10 × PCR buffer, 1mM of each dNTP, 0.5μM of each primer and 2.5U of Taq DNA polymerase (TAKARA, Dalian, China). The DNA was amplified with an initial denaturation of 95 °C for 5 min, followed by 35 cycles of amplification (95°C for 30s, 57°C for 30s, and 72°C for 2min) and a final extension of 72°C for 10min.
Genome cloning and sequencing
The amplified PCR products were separated by electrophoresis on a 1% agarose. The bands were extracted and purified using the AxyPrep DNA Gel Extraction Kit (AxyGene, USA). Then, the PCR products were ligated into the pMD-18T Vector System (Takara Co. Dalian, China), and the recombinant plasmids were sequenced by Takara Co. (Dalian, China).
Phylogenetic analysis
To understand the genetic relationship between the PCV1 isolates from Guangxi, 40 published genomic sequences were downloaded from GenBank (Table 1). All sequences were aligned with Clustal W [19] and were analyzed. The phylogenetic tree was calculated using the Maximum Likelihood (ML) method with 1000 bootstrap replicates and the genetic distance of rep genes, cap genes and complete genomes were calculated using the Kimura 2-parameter, Hasegawa-kishino-Yano and Tamura-Nei model respectively by MEGA6 program [20].
Recombination analysis
Recombination event analysis was carried out by analyzing the complete genome of potential recombinant of GXdx84 since the strain clustered as a separate branch located between PCV1 and PCV2 in the phylogenetic tree based on complete genome as well as cap gene and rep gene. In the recombination events, possible breakpoint were identified using two programs based on different approaches: the RDP4 (http://web.cbio.uct.ac.za/~darren/rdp.html) [21] and SimPlot(http://sray.med.som.jhmi.edu/RaySoft) [22]. The RDP tests the recombination events by six methods (GENECONV, BootScan, MaxChi, Chimaera, SiScan and RDP) and the setting for each method was adjusted account for the dataset features according to the RDP manual recommendations. Recombination events detected by more than 4 methods, where a significance value<10-5 (P-value<10-5) and Bonferroni correction were accepted. The recombination signal and sequences of recombination parental lineages were analyzed by SimPlot. SimPlot analysis was performed with three groups of complete genome: a group of major parent (PCV1G, JN398656), a group of minor parent (PCV2b, AF112862) and a group of potent recombinant (GXdx84, KY437725).
Selection pressure analysis
The selective pressure analysis of genome was assessed by calculating the difference between the dN and dS rates for the aligned rep and cap genes by using MEGA version 6.0 software [23]. The entropy was used to measure the genetic complexity which was calculated by BioEdit [24]. The difference in entropy was plotted between dN and dS [25]. If the dN rate was higher than the dS rate, i.e., dN–dS>0, the genes would be considered to be under positive selection; otherwise, the genes would be considered to be under purified selection (dN–dS<0). If the dN rate was approximately equal to the dS rate, i.e., dN–dS=0, the genes would be considered to be under neutral evolution [26].
Results
Screening for PCV1 prevalence in clinical samples
Clinical samples (n=247) were collected from the lungs, spleen and lymph nodes of apparently healthy pigs (n=172) and sick pigs (n=75) from different regions of Guangxi Province in 2015. Among collected samples, 32 positive samples are PCV1 (positive rate, 12.95%); among these ones, 21 were from the 75 sick pigs (21/75, 28%) and 11 were from the 172 apparently healthy pigs (11/172, 6.39%). Besides of the 247 samples, 214 were PCV2 positive (positive rate 86.6%). 65 samples were from sick pigs (65/75, 86.67%), 149 were from apparently healthy pig (149/172, 86.62%). Both PCV1 and PCV2 were detected in samples from both diseased and apparently healthy pigs.
Phylogenetic analysis of the PCV1 isolates
Fourteen complete genome sequences were randomly selected from the 32 positive samples of PCV1 by PCR (Table 2). Of these samples, 13 strains had a whole-genome length of 1759 nucleotides, and only the GXdx84 strain had a genome length of 1757 nucleotides. Regarding the length of the cap gene, 12 strains had a length of 702 nucleotides, similar to other strains. But the GXyl224 strains had a length of 708 nucleotides (Fig. 1a). As the stop codon TAA mutated to TAT and added Y and K amino acids were added to the end of the Cap protein. This is the first case of PCV1 wherein a structural gene mutation resulted in gene extension. Considering the complete genome, the similarity among the 13 strains (except GXdx84) in this study ranged from 97.8% to 99.6%. In addition, the nucleotide similarity to the other 40 comparison strains of PCV1 ranged from 97.1% to 99.8%. The percentage of mutations were 13.47% (237 mutations) in the whole genome, 16.24% (114 mutations) in cap, and 11.40% (107 mutations) in rep. The nucleotide sequence similarity for the complete gene of GXdx84 strains ranged from 86.8% to 89.0% comparing to PCV2 strains used in this study and 86.8% to 89.0% comparing to the other 40 PCV1 strains. The similarity of rep gene ranged from 92.2% to 94% in PCV1 and 87.6% to 88.3% in PCV2. While the similarity of cap gene ranged from 65.3% to 66.5% in PCV1, and 88.1% to 99.5% in PCV2, especially the similarity to PCV2d-2 was as high as 99.5%. These results indicated that strain of GXdx84 might contain a rep came from PCV1 and a cap came from PCV2.

Alignment of the nucleotide sequence and deduced amino acid for the cap gene, and Cap protein of partial strains analyzed in this study. Conserver residues are indicated by dashed lines. PCV1 is used as the majority sequence for this alignment (AY193712). a The cap gene of GXyl224 inside the red box is one wherein the stop codon mutation led to an increase in the number of nucleotides. b The major nucleotide mutation sites of Cap protein are presented within the red box
We also identified several novel amino acid substitutions in the ORF2 genes from PCV1 isolated in this study. The PCV1 isolates showed 6 amino acid substitutions differed from the PCV1 reference strains: 60 (K to E), 63 (Y to H), 72 (H to Q), 74 (K to R), 176 (H to Q) and 233 (K to E) (Fig. 1b).
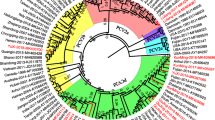
To understand the genetic relationship among the PCV1 isolates in this study, a maximum likelihood (ML) phylogenetic tree was constructed with the 14 strains collected from Guangxi Province, 40 other comparison PCV1 strains, and 4 PCV2 strains with complete genomic nucleotide sequences available in GenBank. Except for the GXdx84 strain, all isolated strains belonged to the PCV1 and showed geographical differences, but these differences were not evident. In particular, the ML phylogenetic tree analysis showed that the GXdx84 strain was different from the other strains (Fig. 2). The GXdx84 strain was located in the branch of neither PCV1 nor PCV2 strains as shown by the whole nucleotide analysis. With regard to the cap and rep gene of the samples, cap of the strain was located in the branch of PCV2, and rep of the strain was located in the branch of PCV1 but was distantly related to other strains of PCV1. These results indicated that the GXdx84 strain might have underwent recombination.

Phylogenetic tree analysis based on the nucleotide sequences of the complete genome (a), cap gene (b), and rep gene (c), performed using the ML method. Numbers along the branches indicate the percentage of confidence in the ML analysis. Only bootstrap support values of >50% are indicated. PCV1 strains isolated in this study are denoted by triangle (▲)
Recombination analysis
The putative recombination events were identified using the Recombination Detection Program (RDP). For 14 PCV1 isolation strains, one recombination event was detected, in which the GXdx84 strain (P-value=1.54×10-13) with the potential parental of PCV1 subtype strain PCV1G and PCV2 subtype strain PCV2b. The number and location of the breakpoints were also determined using similarity plots. When the PCV1G and PCV2b strains were used as potential parental strains, they shared one breakpoint location at 694nt (Fig. 3). It indicated that PCV2b and PCV1G strains might be the minor and major parents of GXdx84 strain.

Simplot analysis the recombination events. One recombination event might be occurred in GXdx84 strain with the PCV1G strain (JN398656) and PCV2b strain (AF112862) as two parent groups. The Y–axis refers to the percentage of similarity. The X–axis refers to the nucleotide position in alignment. The crossed point might be the potential location of recombination event
Selection-pressure analysis of PCV1
The selection-pressure of PCV1 strains was analyzed by calculating the difference in non-synonymous substitution (dN) and synonymous substitution (dS) rates for rep and cap. The differences of dN–dS were -0.09127±0.0251 for the rep gene and -0.2111±0.0541 for the cap gene. These results suggested that the rep gene and the cap gene of PCV1 are under purified selection. Moreover, entropy was coupled with dN–dS to indicate diversity and complexity. The vast majority of Rep and Cap protein amino acid residues exhibited low level or zero entropy (Fig. 4). It indicated that the Rep and Cap protein had low complexity. Only the Cap protein amino acid sequences which were at 50-80, 161-180 and 230-233 had high complexity.

Plot for the difference between non-synonymous and synonymous rates (dN–dS) and amino acid entropy rate for the rep (a) and cap (b) genes
Discussion
PCV1 is considered to be non-pathogenic and economically unimportant and therefore, little is known about its epidemiology and worldwide distribution. More research on PCV1 has focused on the chemic vaccine of PCV1-2 to prevent PCV2 infected in pigs [27, 28]. Recent research found that PCV1 can replicate efficiently and produce pathology in the lungs of porcine fetuses and have a certain impact on porcine alveolar macrophages [16]. It is difficult to rule out the potential damage of PCV1 to the immune system of piglets.
Recombinant events of PCV among different genotypes or different virus isolates have been reported, including recombinant event between PCV2a and PCV2b, and between PCV1 and PCV2a. In this study, we found a recombination GXdx84 strain which was recombinant by PCV1 and PCV2d-2 had a breakpoint at 694nt. Previous research found that the rep gene of the PCV2 can be divided into three regions, and the third region has the highest level of selective pressure which makes the fragment more changeable than the other two [1, 29]. The PCV1 and PCV2 are highly similarity of rep gene and this might be the reason why recombinant event occurred at 694nt.
Most studies on the genetic characterization of PCV2 were based on the cap gene, which is the ideal marker for phylogenetic analysis because this region is considered to be the most variable region in the PCV2 genome and the same phylogenetic tree constructed on the basis of the full genome can be reconstructed with ORF2. However, analysis based on the complete genome is necessary, as it may help identify genetic variability, particularly in terms of recombination events. In this study, genotypic analysis based on the cap gene and complete genome resulted in different phylogenetic trees. The GXdx84 strain was divided into different branches of the phylogenetic tree. Analysis of the cap and rep genes showed that the new strain was clustered in cap in PCV2 and in rep in PCV1. Besides, cap of the GXdx84 strain was highly similar to the genotype of PCV2d-2. In a recent report from China, the positive rate of PCV2d out of PCV2 positive samples ranged from 55% (22/40) [30] to 68.2% (45/66) [31]. It had become the predominant PCV2 subtype [32]. PCV2d can be classified into PCV2d-1 and PCV2d-2, with substantial genetic divergence between the two subtypes. PCV2d-1 strains were first identified in China in 2002 [33], whereas PCV2d-2 strains were first identified in China in 2008 and had been linked with increased virulence [34, 35].
As for the isolated PCV1 strains, the cap gene in the GXyl224 strain spanned 708 nucleotides (codes 235aa), while the other PCV1 strains spanned 702 nucleotides (codes 233aa). This was the first case to report that the capsid protein contains 235 amino acids. Previous studies suggested that the immunoreactive regions of the capsid protein were potential candidate regions involved in the emergence of PCV2 variants [36, 37]. Although PCV1 has no obvious pathogenicity, clinical trials have shown that PCV1 and PCV2 mixed infection generally exhibits the presence of a recombinant strain [38]. Unfortunately, we have not isolated the virus. In the future, we will aim to separate the virus and study its pathogenicity.
In the present study, we also analyzed the selective pressure by calculating the dN and dS rates and the entropy. Very low levels of variability were detected at the nucleotide and amino acid level. Further, the dN–dS values showed that most codons are under neutral or negative selection [39, 40]. Another previous study showed that the average genomic substitution rate for PCV1 was two-fold lower than that for PCV2 [41], which indicated that the PCV1 genome lacked variation. Entropy of Cap and Rep protein coupled with a low complexity, but the Cap protein amino acid sequence which were at 60-80, 161-180 and 230-233 had high complexity (Fig. 4). It was reported that Cap protein was the main antigenic determinant of the PCV2, and contained different epitopes within residues 47-85, 165-200 and the last of the four C-terminal amino acid of the PCV2 capsid protein [42]. Three high complexity regions of PCV1 researched in this study might be the epitopes regions of PCV1. Besides, the Cap protein of GXyl224 coded 235 amino acids, which is two amino acids more than other strains. By analysising the antigenic of GXyl224 by IEDB (http://tools.iedb.org/bcell/), we found that the last six amino acids (230-235aa) were the potential epitope region, other strains of PCV1 were the last four amino acid (230-233aa). Two additional amino acids may result in increased antigenicity of Cap protein, which still need further verification.
Conclusion
In summary, we have sequenced 14 strains suspected to be PCV1 from Guangxi Province, China. ML phylogenetic tree analysis showed that 13 of these strains belonged to PCV1 and 1 strain named GXdx84 belonged neither to PCV1 nor PCV2: it was a chimeric PCV1 and PCV2 strain. In addition, another stain named GXyl224 with ORF2 spans 708 nucleotides by gene sequencing. This is the first study to report that a cap gene mutation resulted in lengthening of in the gene sequence. The correlation between epitope mutations and pathogenicity as well as immunogenicity of GXyl224 and GXdx84 needs further investigation.
Abbreviations
- dN:
-
Non-synonymous substitution
- dS:
-
Synonymous substitution
- ORF:
-
Open-reading frame
- PCV:
-
Porcine circovirus
- PMWS:
-
Post-weaning multi-systemic wasting syndrome
- RDP:
-
Recombination detection program
References
Olvera A, Cortey M, Segales J: Molecular evolution of porcine circovirus type 2 genomes: phylogeny and clonality. Virology 2007, 357(2):175-185.
Cortey M, Segales J. Low levels of diversity among genomes of Porcine circovirus type 1 (PCV1) points to differential adaptive selection between Porcine circoviruses. Virology. 2012;422(2):161–4.
Tischer I, Gelderblom H, Vettermann W, Koch MA. A very small porcine virus with circular single-stranded DNA. Nature. 1982;295(5844):64–6.
Tischer I, Rasch R, Tochtermann G. Characterization of papovavirus-and picornavirus-like particles in permanent pig kidney cell lines. Zentralblatt fur Bakteriologie, Parasitenkunde, Infektionskrankheiten und Hygiene Erste Abteilung Originale Reihe A: Medizinische Mikrobiologie und Parasitologie. 1974;226(2):153–67.
Allan G, Meehan B, Todd D, Kennedy S, McNeilly F, Ellis J, Clark EG, Harding J, Espuna E, Botner A, et al. Novel porcine circoviruses from pigs with wasting disease syndromes. Vet Rec. 1998;142(17):467–8.
Beach NM, Juhan NM, Cordoba L, Meng XJ. Replacement of the replication factors of porcine circovirus (PCV) type 2 with those of PCV type 1 greatly enhances viral replication in vitro. J Virol. 2010;84(17):8986–9.
Cheung AK. Comparative analysis of the transcriptional patterns of pathogenic and nonpathogenic porcine circoviruses. Virology. 2003;310(1):41–9.
Mankertz A, Mueller B, Steinfeldt T, Schmitt C, Finsterbusch T. New reporter gene-based replication assay reveals exchangeability of replication factors of porcine circovirus types 1 and 2. J Virol. 2003;77(18):9885–93.
Cheung AK. Porcine circovirus: transcription and DNA replication. Virus Res. 2012;164(1-2):46–53.
Mankertz A, Caliskan R, Hattermann K, Hillenbrand B, Kurzendoerfer P, Mueller B, Schmitt C, Steinfeldt T, Finsterbusch T. Molecular biology of Porcine circovirus: analyses of gene expression and viral replication. Veterinary microbiology. 2004;98(2):81–8.
Nawagitgul P, Morozov I, Bolin SR, Harms PA, Sorden SD, Paul PS. Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J Gen Virol. 2000;81(Pt 9):2281–7.
Bratanich AC, Blanchetot A. PCV2 replicase transcripts in infected porcine kidney (PK15) cells. Virus Genes. 2002;25(3):323–8.
Chaiyakul M, Hsu K, Dardari R, Marshall F, Czub M. Cytotoxicity of ORF3 proteins from a nonpathogenic and a pathogenic porcine circovirus. J Virol. 2010;84(21):11440–7.
Tischer I, Bode L, Peters D, Pociuli S, Germann B. Distribution of antibodies to porcine circovirus in swine populations of different breeding farms. Arch Virol. 1995;140(4):737–43.
Rodriguez-Arrioja GM, Segales J, Balasch M, Rosell C, Quintant J, Folch JM, Plana-Duran J, Mankertz A, Domingo M. Serum antibodies to porcine circovirus type 1 and type 2 in pigs with and without PMWS. Vet Rec. 2000;146(26):762–4.
Saha D, Lefebvre DJ, Ducatelle R, Doorsselaere JV, Nauwynck HJ. Outcome of experimental porcine circovirus type 1 infections in mid-gestational porcine foetuses. BMC Vet Res. 2011;7:64.
Calsamiglia M, Segales J, Quintana J, Rosell C, Domingo M. Detection of porcine circovirus types 1 and 2 in serum and tissue samples of pigs with and without postweaning multisystemic wasting syndrome. Journal of clinical microbiology. 2002;40(5):1848–50.
Fenaux M, Opriessnig T, Halbur PG, Xu Y, Potts B, Meng XJ. Detection and in vitro and in vivo characterization of porcine circovirus DNA from a porcine-derived commercial pepsin product. The Journal of general virology. 2004;85(Pt 11):3377–82.
Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic acids research. 1994;22(22):4673–80.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution. 2011;28(10):2731–9.
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26(19):2462–3.
Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. Journal of virology. 1999;73(1):152–60.
Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21(5):676–9.
Wolinsky SM, Korber BT, Neumann AU, Daniels M, Kunstman KJ, Whetsell AJ, Furtado MR, Cao Y, Ho DD, Safrit JT. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science. 1996;272(5261):537–42.
Zhang Z, Jia R, Lu Y, Wang M, Zhu D, Chen S, Yin Z, Chen X, Cheng A. Identification, genotyping, and molecular evolution analysis of duck circovirus. Gene. 2013;529(2):288–95.
Hughes AL, Nei M: Nucleotide substitution at major histocompatibility complex class II loci: evidence for overdominant selection. Proceedings of the National Academy of Sciences of the United States of America 1989, 86(3):958-962.
Seo HW, Oh Y, Han K, Park C, Chae C. Reduction of porcine circovirus type 2 (PCV2) viremia by a reformulated inactivated chimeric PCV1-2 vaccine-induced humoral and cellular immunity after experimental PCV2 challenge. BMC Vet Res. 2012;8:194.
Han S, Xiao Y, Zheng D, Gu Y, Xuan Y, Jin Y, Pang W, Huang Y, Li X, Deng J, et al. Establishment and application of a competitive enzyme-linked immunosorbent assay differentiating PCV2 antibodies from mixture of PCV1/PCV2 antibodies in pig sera. BMC Vet Res. 2016;12(1):175.
Cheung AK. Identification of an octanucleotide motif sequence essential for viral protein, DNA, and progeny virus biosynthesis at the origin of DNA replication of porcine circovirus type 2. Virology. 2004;324(1):28–36.
Cai L, Ni J, Xia Y, Zi Z, Ning K, Qiu P, Li X, Wang B, Liu Q, Hu D, et al. Identification of an emerging recombinant cluster in porcine circovirus type 2. Virus Res. 2012;165(1):95–102.
Wei C, Zhang M, Chen Y, Xie J, Huang Z, Zhu W, Xu T, Cao Z, Zhou P, Su S, et al. Genetic evolution and phylogenetic analysis of porcine circovirus type 2 infections in southern China from 2011 to 2012. Infect Genet Evol. 2013;17:87–92.
Perez LJ, Diaz de Arce H, Frias MT. Genetic characterization and phylogenetic analysis of porcine circovirus type 2 strains present in Cuban swine herds. Res Vet Sci. 2010;89(2):301–5.
Cortey M, Olvera A, Grau-Roma L, Segales J. Further comments on porcine circovirus type 2 (PCV2) genotype definition and nomenclature. Veterinary microbiology. 2011;149(3-4):522–3.
Guo LJ, Lu YH, Wei YW, Huang LP, Liu CM. Porcine circovirus type 2 (PCV2): genetic variation and newly emerging genotypes in China. Virology journal. 2010;7:273.
Xiao CT, Halbur PG, Opriessnig T. Global molecular genetic analysis of porcine circovirus type 2 (PCV2) sequences confirms the presence of four main PCV2 genotypes and reveals a rapid increase of PCV2d. J Gen Virol. 2015;96(Pt 7):1830–41.
Lv QZ, Guo KK, Zhang YM. Current understanding of genomic DNA of porcine circovirus type 2. Virus genes. 2014;49(1):1–10.
Larochelle R, Magar R, D'Allaire S. Genetic characterization and phylogenetic analysis of porcine circovirus type 2 (PCV2) strains from cases presenting various clinical conditions. Virus Res. 2002;90(1-2):101–12.
Gagnon CA, Music N, Fontaine G, Tremblay D, Harel J. Emergence of a new type of porcine circovirus in swine (PCV): a type 1 and type 2 PCV recombinant. Vet Microbiol. 2010;144(1-2):18–23.
Csagola A, Kiss I, Tuboly T. Detection and analysis of porcine circovirus type 1 in Hungarian wild boars: short communication. Acta veterinaria Hungarica. 2008;56(1):139–44.
Muhling J, Raye WS, Buddle JR, Wilcox GE. Genetic characterisation of Australian strains of porcine circovirus types 1 and 2. Australian veterinary journal. 2006;84(12):421–5. discussion 416
Firth C, Charleston MA, Duffy S, Shapiro B, Holmes EC. Insights into the evolutionary history of an emerging livestock pathogen: porcine circovirus 2. Journal of virology. 2009;83(24):12813–21.
Lekcharoensuk P, Morozov I, Paul PS, Thangthumniyom N, Wajjawalku W, Meng XJ. Epitope mapping of the major capsid protein of type 2 porcine circovirus (PCV2) by using chimeric PCV1 and PCV2. J Virol. 2004;78(15):8135–45.
Acknowledgements
We would like to thank the native English staff of Elixigen Company (Huntington Beach, California) for editing the manuscript. And we would like to thank Ningning Hu for her assistance with reagent preparation.
Funding
This study was funded by the National Program on Key Research Project of China [grant number 2017YFD0500101 and 2016YFD0500401] and the National Natural Science Foundation of China [grant number 31272573].
Availability of data and materials
The datasets used and/or analyzed during the current study is available from the corresponding author on reasonable request. The dada from this study is also available from TreeBASE, http://purl.org/phylo/treebase/phylows/study/TB2:S22117.
Author information
Authors and Affiliations
Contributions
LC performed the research, data analysis and wrote the manuscript. WCS, HJL and MYT helped in the writing of the manuscript. CZX, GYZ, JCH, WW and MZ participated in sample collection and research testing. RD, NYJ and ADQ designed the research. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Experimental protocols for obtaining pigs clinical samples used in this study were obtained from Guangxi Center for Animal Disease Control and Prevention and carried out in strict accordance with the Animal Ethics Procedures and Guidelines of the People's Republic of China. All pigs were euthanized by an anesthetic overdose with the pentobarbital before collected the samples and all pig owners agree the samples’ usage before participation in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cao, L., Sun, W., Lu, H. et al. Genetic variation analysis of PCV1 strains isolated from Guangxi Province of China in 2015. BMC Vet Res 14, 43 (2018). https://doi.org/10.1186/s12917-018-1345-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-018-1345-z