
Abstract
Background
Much of the structure-based mechanistic understandings of the function of SLC6A neurotransmitter transporters emerged from the study of their bacterial LeuT-fold homologs. It has become evident, however, that structural differences such as the long N- and C-termini of the eukaryotic neurotransmitter transporters are involved in an expanded set of functional properties to the eukaryotic transporters. These functional properties are not shared by the bacterial homologs, which lack the structural elements that appeared later in evolution. However, mechanistic insights into some of the measured functional properties of the eukaryotic transporters that have been suggested to involve these structural elements are sparse or merely descriptive.
Results
To learn how the structural elements added in evolution enable mechanisms of the eukaryotic transporters in ways not shared with their bacterial LeuT-like homologs, we focused on the human dopamine transporter (hDAT) as a prototype. We present the results of a study employing large-scale molecular dynamics simulations and comparative Markov state model analysis of experimentally determined properties of the wild-type and mutant hDAT constructs. These offer a quantitative outline of mechanisms in which a rich spectrum of interactions of the hDAT N-terminus and C-terminus contribute to the regulation of transporter function (e.g., by phosphorylation) and/or to entirely new phenotypes (e.g., reverse uptake (efflux)) that were added in evolution.
Conclusions
The findings are consistent with the proposal that the size of eukaryotic neurotransmitter transporter termini increased during evolution to enable more functions (e.g., efflux) not shared with the bacterial homologs. The mechanistic explanations for the experimental findings about the modulation of function in DAT, the serotonin transporter, and other eukaryotic transporters reveal separate roles for the distal and proximal segments of the much larger N-terminus in eukaryotic transporters compared to the bacterial ones. The involvement of the proximal and distal segments — such as the role of the proximal segment in sustaining transport in phosphatidylinositol 4,5-bisphosphate-depleted membranes and of the distal segment in modulating efflux — may represent an evolutionary adaptation required for the function of eukaryotic transporters expressed in various cell types of the same organism that differ in the lipid composition and protein complement of their membrane environment.
Similar content being viewed by others


Background
The dopamine transporter (DAT) is a member of the neurotransmitter:sodium symporter (NSS) family of proteins belonging to the solute carrier 6 (SLC6) family that performs the reuptake of dopamine from the synaptic cleft into the presynaptic nerve required for neuronal signaling [1]. The essential role of DAT, and of its closely related homologs — the serotonin and norepinephrine transporters (SERT and NET) — in signal termination at the synapse makes them important targets for psychostimulants such as cocaine and amphetamines, as well as for pharmaceutical treatment of a variety of disorders of the nervous system [2]. Moreover, genetic modifications of the functions of these transporters [3, 4] have been implicated in diseases including schizophrenia, Parkinson’s disease, and attention-deficit/hyperactivity disorder (ADHD). Reverse transport of the neurotransmitters (efflux) mediated by DAT and SERT, which has been shown to be affected by such disease-related mutations, is currently a very active topic of research on mechanisms of these membrane proteins [4,5,6,7,8,9,10,11,12,13,14,15].
Much has been learned about these mammalian neurotransmitter transporters from the investigation of the structure and function of their bacterial homologs, with which they share many structural and mechanistic properties [16,17,18,19]. It has become clear, however, that important structural differences exist between the eukaryotic and bacterial proteins, the largest being the much longer N- and C-termini that have been proposed to be partially structured [20, 21]. Notably, experimental data point to an involvement of these regions of structural difference in measured functional properties of the mammalian NSS [22,23,24]. For example, the phosphorylation of the N-terminus has been implicated in the efflux functions of the human DAT (hDAT) [22, 25], and our work has shown that the amphetamine (AMPH)-induced reverse transport (efflux) exhibited by DAT and SERT, but not by the bacterial analogs, is dependent on electrostatic interactions between the hDAT N-terminus and negatively charged phosphatidylinositol 4,5-bisphosphate (PIP2) lipids in the membrane [34, 41]. Notably, the measured substrate uptake by DAT, a function it shares with the bacterial transporters, is not affected by this N-terminus interaction [34].
Structure-based mechanistic details of the mode in which eukaryotic NSS function is modulated by the involvement of the N-terminus are still sparse, but mechanistic insight from computational studies of hDAT has shown [29] that the N-terminus of DAT engages the PIP2 lipid component of membranes to achieve conformational changes related to function (CCRF). These include (1) the transition of the DAT from outward-facing to inward-facing configurations [30] and (2) the release of the sodium ion from the Na2 binding site that is known to precede substrate transport [26, 31] and can serve as a monitor of the initial stages of the functional mechanism [32]. The complexity of the different CCRF suggested that the N-terminus/PIP2 interactions supporting them may (1) involve different modes of association with the membrane under various conditions (such as in the presence/absence of PIP2) and (2) would be different for different hDAT constructs (e.g., when the N-terminus is phosphorylated or mutated). This reasoning provided a specific testable hypothesis about the mechanism by which new properties of the eukaryotic transporters, those that are not shared with the bacterial homologs, are enabled by the addition of the long N- and C-terminals.
To probe this hypothesis and verify the relation between the experimental measurements and the specific modes in which the N-terminus participates, we undertook the present computational study of the modes of interaction of the N-terminus in hDAT with the rest of the structure, including the C-terminus, under various conditions and with modifications (phosphorylation, mutations) that have known functional consequences. Here we describe the results from extensive ensemble-level all-atom molecular dynamics simulations we used in this study that also included Markov state model (MSM) analysis of hDAT dynamics modulated by mutated and/or modified N-terminus constructs and conditions. The results reveal preferred modes of interaction of the N-terminus with the intracellular domains of hDAT, which can be directly associated with experimentally measured functional phenotypes of the transporter. We show how these interaction patterns change under conditions that have been demonstrated to selectively affect efflux but not regular transport, e.g., PIP2 depletion, mutations such as R51W, the K3A/K5A double mutation, or phosphomimic substitution, S/D, of serine residues at positions 2, 4, 7, 12, and 13 to aspartate [22, 33, 34]. Moreover, we verify, for the first time, the consistency of observations relating quantitative measures of the specific modes of interaction of the N-terminus with the measured functional properties attributed to them.
The coherent and direct relation between experimentally determined effects of the mutations and conditions and the interaction modes identified from the simulations validates the computational results and mechanistic conclusions. Moreover, because the mechanistic inferences are described in atomistic detail, they offer specific experimentally testable predictions for further studies of SLC6 transporter function and of the structure-based relation between the function of bacterial and eukaryotic members of this family. In particular, the details of the rich spectrum of modes of interaction of the long N-terminus of hDAT that emerges from these studies reveal the different roles of the distal and proximal segments of the N-terminus in modulating specific functions of hDAT. As these are segments of the much larger N-terminus shared by the eukaryotic transporters compared to the bacterial ones, the findings bring mechanistic support for our proposal that the size of this region increased during evolution so as to enable more, and different, modes of regulation that are not shared with the bacterial analogs. An example discussed in detail is the mechanistic explanation for experimentally determined differences in the effects on uptake vs efflux resulting from manipulation of the N-terminus by partial truncation, mutations, and/or elimination of PIP2 interactions. This example further underscores the central role of this structural addition in the evolution from the bacterial LeuT-like members of this family.
Results
The full complement of molecular dynamics (MD) simulation trajectories carried out specifically for this study, as described in Methods, includes 50 statistically independent ~ 1-μs-long trajectories for each of the four different conditions and constructs for which quantitative measurements of activity are available. As discussed before [32], the ensemble exploration of the configurational space of each construct/condition bolsters the statistical validity of the inferences and predictions from the simulation. The specific molecular systems investigated in this manner include (1) wild-type hDAT in PIP2-depleted membranes (hereafter referred to as the “no-PIP2 system”), (2) hDAT with the R51W mutation in the N-terminal domain, with the double K3A + K5A mutation in the N-terminus (termed the “K3/5A system”), and those with the first five N-terminal serine residues substituted by Asp as a phosphomimic (termed “S/D construct”); these three mutant constructs were immersed in PIP2-containing bilayers, consistent with the experimental conditions under which their functional properties were assayed. The data from these computations are compared to results for the wild-type hDAT simulated in PIP2-containing membranes we reported recently [32], and are analyzed utilizing the same protocols as described therein and detailed here in Methods.
Different modes of interaction of the N-terminus with the rest of the hDAT protein correspond to differences in experimentally measured functional properties
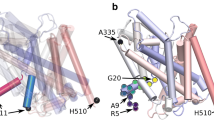
From the simulation trajectories we identified the regions of hDAT structure that interact with the N-terminus, and generated the per-residue contact map shown in Fig. 1. This map shows that with PIP2 present in the membrane, the N-terminus, as a whole, interacts with all intracellular loop (IL) regions of hDAT (IL1, IL2, IL3, IL4, and IL5), and with the C-terminus.

hDAT structure and N-terminus interactions. (Left) Contact map for interaction of N-terminus residues (x-axis) with all hDAT residues (y-axis). Distance calculations were done with the closest-heavy algorithm implemented in MDTraj software [83]. The color bar shows the minimum distance of each residue in the N-terminus to other residues (the lightest colored regions indicate the strongest interactions, e.g., in the various loop segments) as identified in the ensemble of 50-μs trajectories for the wild-type hDAT embedded in the PIP2-containing lipid bilayer (see Methods). (Right) Snapshot of hDAT structure highlighting the intracellular segments interacting with the N-terminus in the ensemble of 50-μs trajectories. Charged residues are shown in licorice
A detailed comparative analysis of the interactions between the N-terminus with the intracellular regions of wild-type hDAT in PIP2-containing membranes (obtained from equivalent trajectories described recently [32]) and the constructs studied here (including PIP2-depleted membrane conditions) reveals a specific pattern (modes) of interaction of the different parts of the N-terminus with intracellular regions of the transporter. These patterns are presented in Fig. 2.

Modes of interaction of hDAT N-terminus with intracellular loop segments. The bars show the average interaction strength calculated from all 50 trajectories of every construct as described in Methods. Error bars show standard deviations (see Methods for full details of calculations and error estimation)
As part of the experimental studies of reverse substrate transport (efflux) by DAT [22] and in SERT [35], their N-terminus was truncated, in the case of DAT eliminating the first 22 residues (∆N22 system). The measurements showed that the truncated transporters maintain direct substrate transport (uptake), but efflux is severely impaired. To compare with and interpret these experimental results, we calculated the modes of interaction of the various constructs for the corresponding components of the N-terminus: the distal N-terminus (residues 1–22) and the proximal N-terminus (residues 23–57). The results in Fig. 2 show a distinct difference between the interaction patterns of the two segments. Notably, the largest differences are registered for the interactions of these distal and proximal segments with IL1, IL4, and the C-terminus. A remarkable similarity is revealed between the pattern of interactions with IL4 calculated for the entire N-terminus, and for just the proximal N-terminus (cf. the circled regions in Fig. 2). This similarity is especially noteworthy because the interaction with IL4 has been singled out to be essential in the early steps of the substrate transport mechanism marked by the release of Na+ from the Na2 site [29]. Thus, our finding here that the proximal N-terminus maintains the essential pattern of interaction with IL4 explains the surprising insensitivity of the inward substrate transport to the deletion of the first 22 residues.
The second largest difference between the interaction patterns of the proximal and distal N-terminal segments evident in Fig. 2 pertains to association with the C-terminus. In particular, the distal segment of the phosphomimic S/D construct has a remarkably high interaction quotient with the C-terminus, whereas the weakest association with the C-terminus is observed for the K3/5A construct (in fact, the distal segment of this double mutant K3A/K5A N-terminus is seen to engage in the least amount of interactions with any of the intracellular loops). This diametrically opposed characteristic of the interaction of the two constructs relative to the wild type is remarkable and is fully consistent with the experimentally determined functional properties compared to the wild-type hDAT: (1) only the S/D mutant construct in which the distal N-terminus interacts strongly with the C-terminus has been found thus far to be able to enhance dopamine efflux mediated by DAT in the absence of AMPH [22], and (2) the K3/5A mutant, which we find to have the weakest interaction between the distal segment and the C-terminus, produces a very low AMPH-induced dopamine efflux [34]. The important relation of the distal segment with the efflux phenotype is underscored by the deleterious effect of the (∆N22) truncation on efflux, but not on uptake, as discussed above.
Multiple paths of inward release of Na+ from the Na2 site are regulated by the modes of interaction of the N-terminus
Our detailed study of the release of Na+ from the Na2 site (termed Na+/Na2 release) — which is known to initiate solute translocation by the hDAT [26, 31] — has identified CCRF and the underlying allosteric mechanism [27, 28, 32]. These CCRF relate directly to the mode of interaction of the N-terminus with intracellular regions of the transporter in PIP2-containing membranes [32]. In the earlier studies [29, 32] we showed that specific PIP2-mediated associations between the N-terminus and various intracellular loop regions of DAT trigger conformational transitions related to the release of Na+/Na2. Here we find, from the new sets of MD simulations of the mutant constructs and conditions we study, that while they differ in their modes of N-terminus interactions (Fig. 2), Na+/Na2 release is observed, albeit at different rates, during simulations of the same time length for various systems (Additional file 1: Figures S1 to S4). While the rates of release events observed in the 50 trajectory ensembles for each construct/condition (see Additional file 1: Figures S1 to S4) are not rigorously comparable to each other in a statistically meaningful manner, the prediction of inward release of Na+/Na2 in each of these constructs is consistent with experimental results and with our previous finding [32] that the destabilization of Na+/Na2 is highly correlated with the amount of water penetration to the binding site (Additional file 2: Table S4). Additional file 1: Figures S5, S6, and S7 show details of the spontaneous release dynamics calculated for the S/D system (Additional file 1: Figure S5), the R51W hDAT system (Additional file 1: Figure S6), and the no-PIP2 system (Additional file 1: Figure S7). The K3/5A mutant did not exhibit a release event, but the pattern of Na+/Na2 destabilization and intracellular gate opening is similar to that observed for S/D (Additional file 1: Figures S3, S4, S8), suggesting that it is on the path to Na+/Na2 release as well.
These results for the large number of different constructs and conditions are remarkably consistent with the experimental evidence showing that the regular transport of the dopamine substrate (uptake) is affected differently by the various mutations/conditions than the reverse transport of this substrate (efflux) induced by AMPH. Thus, efflux is impaired by most of these mutations/conditions, with the exception of the S/D system, which exhibits dopamine efflux even in the absence of AMPH but under elevated intracellular Na+ concentrations [25]. In particular, experimental evidence points to the importance of PIP2-containing membranes for various functional phenotypes of hDAT, including AMPH-induced efflux, but shows that substrate transport is only mildly affected if PIP2 content is reduced [34,35,36].
The dependence of functional properties of the eukaryotic transporters on PIP2 is not shared by the bacterial transporter homologs, such as the structural prototype LeuT [16, 37], which do not require PIP2-containing membranes for transport and also do not exhibit reverse transport. We reasoned that comparing molecular details of functional mechanisms involving the N-terminus in the presence and absence of PIP2 would shed new light on the role introduced in evolution by the long N-terminus of the eukaryotic transporters. To discern the source of underlying mechanistic differences that connect PIP2 sensitivity to the long N-terminus, it therefore became necessary to understand (1) how the initiating step of substrate transport, i.e., the release of Na+/Na2, is achieved in PIP2-containing vs PIP2-depleted membranes, and (2) what the role of the N-terminus interactions is in the CCRF (including Na+/Na2 release process) when PIP2 is not present. To this end we used the MSM analysis to obtain a kinetic model for the Na+/Na2 release process in the no-PIP2 system, as the comparison of quantitative terms for the wild-type protein with/without PIP2 allows robust mechanistic inferences as illustrated below.
Markov state model analysis of Na+/Na2 release in PIP2-depleted membranes
To enable direct comparison of the results with the MSM analysis of wild-type hDAT in PIP2-containing membranes [32], we built and analyzed the MSM for the no-PIP2 system following the same protocol (see Methods and Ref. [32]). Thus, as the same mechanism was followed in the two compared conditions (i.e., with/without PIP2), the same set of parameters as before [32] (Additional file 2: Table S3) was used to generate the reduced conformational space with the time-structure-based independent component analysis (tICA) method (see Methods). The tICA energy landscape (Fig. 3b) was obtained by projecting all the conformations from all the trajectories onto the first two tICA reaction coordinates. Visualization of conformations belonging to different regions of the tICA energy landscape revealed that this landscape, unlike the one for PIP2-containing membrane conditions (shown in Fig. 3a), could be divided into only two (rather than three) regions in terms of the location of the Na+/Na2 ion: one in which Na+/Na2 is still bound in the Na2 site, and the other in which Na+/Na2 is already released (Fig. 3b). Thus, this tICA space (Fig. 3b) does not contain a region representing the intermediate state seen in the wild-type hDAT system in PIP2-enriched membranes when Na+/Na2 has left the binding site but is not yet released to the intracellular environment because it is interacting with the E428 side chain (Fig. 3c). Because this interaction requires the E428 side chain to be free from its partner in the E428–R445 gate [32], the results suggest that a change in N-terminus interactions due to PIP2 depletion directly affects this gate. Indeed, the finding summarized in Fig. 2 shows a major reduction in the interactions of the N-terminus with IL4 in the absence of PIP2, which is thus seen to result in a more stable R445–E428 gate in the absence of PIP2 (Fig. 3d, see also Additional file 1: Figure S9).

hDAT tICA space in presence and absence of PIP2 lipids. a and b Population-weighted tICA landscape for hDAT trajectories in PIP2-containing membranes (a) and for the no-PIP2 system (b). For each system, all conformations in all 50 trajectories are projected on the space of the tICA first and second eigenvectors. Regions differing with respect to the location of Na+/Na2 are highlighted on the tICA landscape with transparent boxes of different colors. c All the conformations are projected on the reaction coordinate based on Na+/Na2 distance from the sodium at site Na1, and the free energy (∆G) is population-based and is calculated as [∆G = −RT ln(population)]. The intermediate state is highlighted by the magenta box; note the absence of a distinct minimum in the absence of PIP2 (red line). Error bars (shown as transparent shades) are calculated using the bootstrap method on 100 blocks of frames with 160-ns time range that are randomly extracted from all 50 trajectories. d Comparing percentage of broken interactions of intracellular gates in the control simulation (PIP2-containing membrane) (red bars) and in the no-PIP2 system (blue bars) averaged over all 50 trajectories of each construct. Error bars are calculated using the bootstrap method by randomly selecting 50 trajectories (with replacement) and repeating the process for 1000 iterations
To build the MSM from the two-dimensional (2D) tICA landscape shown in Fig. 3b, we followed the same protocol as before [32] to obtain the implied time-scales plot (see Methods). As shown in Additional file 1: Figure S10 (top panel), Markovian behavior of the system is observed after a lag time of ~ 80 ns; therefore, the transition probability matrix (TPM) (see Methods) at an 80-ns lag time was selected for all subsequent analyses of the no-PIP2 system. Mapping all microstates on the tICA landscape and coloring them based on the first MSM relaxation mode (i.e., the second TPM eigenvector, shown in Additional file 1: Figure S11) reveals that Na+/Na2 is still bound in microstates with positive sign (red circles in Additional file 1: Figure S11), whereas the microstates with negative sign (blue circles in Additional file 1: Figure S11) have released Na+/Na2 to the intracellular environment. Since the state population flows from positive to negative states, the first MSM relaxation mode is seen to capture the overall release kinetics of Na+/Na2. The implied time-scale equation (see Methods) shows that this relaxation mode is characterized by timescales of ~ 1.1 μs, comparable to the previously reported kinetics for hDAT in the PIP2-containing membranes (~ 800 ns) [32] (Additional file 1: Figure S10).
To compare the mechanisms of sodium release from the Na2 site of the wild-type hDAT in PIP2-containing vs PIP2-depleted membranes, we used the same transition path theory (TPT) analysis (see Methods) to obtain the most probable release pathways of Na+/Na2, and quantified the flux associated with each of these on a macrostate-based MSM using 15 macrostates as before [32]. Similar to the wild type in PIP2-containing membranes, several pathways are revealed in the no-PIP2 system. Here, the first 10 pathways identified by the TPT analysis contribute ~ 80% of the total flux between Na+/Na2 bound states and Na+/Na2 released states (highlighted in Fig. 4; see Additional file 2: Table S5 for quantification of fluxes). Their structural context is shown in Fig. 4 and Additional file 1: Figure S13.

Comparison of Na+/Na2 release pathways calculated in PIP2-enriched and PIP2-depleted membranes. Results from the transition path theory (TPT) analysis for the release of Na+/Na2 to the intracellular environment by the wild-type hDAT in PIP2-containing membrane (top) and in PIP2-depleted membrane (bottom) systems. Arrow thickness represents relative flux magnitude for each transition. The numbers above circles show corresponding macrostate numbers. A representative conformation is shown for selected macrostates, with IL2 highlighted in orange (cartoon representation) on the transparent structure of hDAT. Density representations of the residues forming intracellular gates are shown as fuzzy background gray colors and the most probable locations of these residues within the density representation are highlighted in licorice rendering and labeled for reference. Dopamine is shown in pink licorice and sodium and chloride ions in yellow and cyan spheres, respectively. Red, green, and blue boxes highlight the three major release pathways of Na+/Na2; numbers indicate the fraction of the total flux carried by each major pathway
The identification of the major Na+/Na2 release pathway in the no-PIP2 system reveals a clear difference from the release in the PIP2-enriched system (cf. panels labeled “with PIP2” and “without PIP2” in Fig. 4) that echoes the difference in the modes of interaction described by the results in Fig. 2. In the no-PIP2 system, the first major pathway for the release contributes ~ 40% of the total flux and is formed when both the R60–D436 and the E428–R445 gates remain closed (a consequence of the much lower interaction with IL4 as seen in Fig. 2), but the K66–D345 is open (highlighted in the transparent red box in Fig. 4). This is in sharp contrast to the result in the presence of PIP2 where this pathway is a minor contributor to the total flux, at only ~ 13%.
The second major pathway in the no-PIP2 system is enabled when both the R60–D436 and E428–R445 gates are broken, but R60 is now able to form interactions with E428 (Fig. 4, green box). This pathway contributes ~ 32% to the total flux. Notably, this was the major Na+/Na2 release pathway in the presence of PIP2, contributing about 48% percent of the total flux. But because in the absence of PIP2 R60 (or R445) is interacting with E428, the intermediate state cannot form.
The third major pathway in the no-PIP2 system is formed when the R60–D436 gate is broken and the N-terminus moves closer to IL2. The intracellular gate interaction E428–R445 is still maintained (Fig. 4, blue box). In the presence of PIP2, this pathway contributes about ~ 39% to the total flux, but in the no-PIP2 system the contribution is reduced to 28%. Importantly, a major difference is that in the presence of PIP2, R60 is seen to be engaged in PIP2-mediated interactions with several positively charged residues from IL2 (K257, K260, K264), whereas in the no-PIP2 system the association between the N-terminus and the IL2 region still permits interactions between R60 and D68 and/or D345 (see macrostate 3 in Fig. 4 and Additional file 1: Figure S13).
Overall, the TPT analysis shows that the preference ranking of the various release pathways, which we quantified by calculating fluxes, is strongly affected by the PIP2 lipids in a manner consistent with the modes of interaction with the N-terminus. The difference in Na+/Na2 mechanism between the two conditions (with/without PIP2) is underscored by the finding that the increased dynamics of the N-terminus (Additional file 1: Figure S12) in the absence of PIP2 results in a destabilization of the K66–D345 gate, which enhances the Na+/Na2 release flux through a pathway that was only marginally active in the presence of PIP2 (pathway highlighted in red box). Taken together, these results show how the eukaryotic transporters can adapt to different membrane composition conditions by utilizing different N-terminus interaction patterns so that release of Na+ from the Na2 site is maintained.
Discussion
The extensive investigations of the molecular mechanisms underlying the vital role of hDAT in signal transduction have profited much from the availability of structurally simpler prototypes of the NSS family, such as the bacterial analog LeuT (a leucine and alanine transporter), for which the crystallographic data provided the first structural basis for detailed molecular studies (see [16, 37]). When the striking fold similarity of LeuT to the eukaryotic and human neurotransmitter transporters such as DAT was established [38, 39], it enabled major progress in understanding the functional mechanism, as documented in a very large number of publications (for reviews see [40]). It became clear, however, that various physiologically important mechanisms that eukaryotic transporters such as DAT and SERT have acquired through evolution involve allosteric coupling to their environment that differentiate them from the structurally simpler bacterial analogs. In a large number of studies [20, 22, 24, 25, 29, 32, 34, 41,42,43,44,45,46,47,48,49,50] to which we have contributed findings from both computation and experiments, these new functions of the eukaryotic transporters — such as regulation by lipids and by phosphorylation, and the observed efflux phenotype — were shown to be mechanistically modulated by defined structural elements, especially the relatively long N- and C-terminal segments that are found exclusively in this class, and not in bacterial counterparts [39]. These intracellularly located N- and C-terminal segments are the most divergent structural units within the NSS family of proteins, as they range in size from just a few (~ 10) amino acids (in bacterial members, such as LeuT) to segments containing > 60 residues (N-terminus of eukaryotic NSS such as DAT, SERT; see [39, 51]).
To understand how the new functions of the eukaryotic transporters relate to the presence of the N-terminus and its dynamic properties, we quantified the interaction of the hDAT N-terminus with the intracellular regions of the hDAT transporter. On this basis we were able to relate different modes of interaction to the functional mechanisms of DAT through the relation between the experimentally measured effects of mutations/conditions and the changes in modes of interaction. The release of the Na+ ion from the Na2 site, which is known to be a key first step in the substrate translocation cycle [26, 31], served as an established functional readout for the initiation of the transport cycle. We showed how the effects of modes of interaction of the N-terminus with various intracellular regions of the transporter relate to various modes of intracellular gate opening and paths of water penetration. This led to a mechanistic interpretation of the experimentally measured modifications of the functional properties observed for mutant constructs in the presence and absence of PIP2. Together, our results reveal how changes in the modes of interaction stemming from N-terminus mutations and PIP2 depletion are directly associated with stabilization/destabilization of intracellular gates, and also show their effect on the penetration of water into the binding site (Fig. 5a) that is required for uptake and efflux in hDAT.

Illustration of effects of different modes of N-terminus interactions on functional phenotypes. a Representation of two water penetration pathways from the intracellular side of hDAT. The N-terminus/IL4 water pathway is the main water penetration path in the wild type in the presence of PIP2 lipids. In the absence of PIP2, the N-terminus/IL4 water (blue) penetration pathway is impaired, and hydration of the binding site is achieved primarily through another water pathway formed between TM1a and IL3 (red). b Representation of conditions linked to the efflux function of hDAT. The interaction of the proximal N-terminus with IL4 (blue) is mediated by PIP2 lipids, and the distal part of the N-terminus engages with the C-terminus (red). All charged residues are shown for both N-terminus and C-terminus with thin licorice; charged residues engaged in interactions between N-terminus and either IL4 or the C-terminus are highlighted with thick licorice. Dopamine is shown in the S1 binding site (in pink), sodium ions are shown as yellow spheres, and the chloride ion is in cyan sphere
By specifying the modes of interactions of the N-terminus that regulate the opening probabilities of water pathways separately for the distal and the proximal segments of the N-terminus, our findings provide a clear mechanistic explanation for a persistent conundrum in the literature regarding the effects of (1) truncating the first 22 residues of the N-terminus, and (2) PIP2 depletion, and in particular their different effects on the uptake and efflux aspects of eukaryotic transporter function. That the truncation has little effect on uptake is explained by our finding (summarized in Fig. 2) that the pattern of interactions of the N-terminus with IL4 of DAT is mimicked by just the proximal segment, without special involvement of the distal segment. However, this interaction, which has been shown to disrupt the E428–R445 gate and thus serves as one of the major mechanistic triggers for Na+/Na2 release, is PIP2-mediated [29] and is reduced under no-PIP2 conditions (Fig. 2). This might have suggested a reduced release of Na+/Na2 that would impair the substrate uptake function under these conditions. Yet this is not what is observed experimentally, as the uptake function of hDAT is not impaired by PIP2 depletion from the membrane. The mechanistic explanation emerges from our analysis of (1) the simulations presented here that show how modes of interaction of the N-terminus change under the different conditions (and for the various mutant constructs; Fig. 2), and (2) the MSM from the simulation trajectories of the no-PIP2 condition showing how Na+/Na2 release is maintained, but with different probabilities of the release pathways that involve alternative water penetration channels (e.g., the TM1a/IL3 channel (Fig. 5a) that is formed when K66–D345 interaction breaks).
The effect of N-terminus truncation on efflux is even more interesting in the context of the functional difference between the bacterial and eukaryotic transporters, because elimination of the distal segment in the neurotransmitter SLC6 transporters impairs a function not shared with the bacterial homologs. The relation of the distal segment to efflux makes it tempting to speculate that some combination of the interaction modes of the distal and proximal N-terminus (Fig. 5b) is required for efflux. Indeed, the results summarized in Fig. 2 show (1) a reduction of proximal N-terminus/IL4 interactions in constructs with low efflux activity (R51W and K3/5A) and in PIP2-depleted membranes and (2) a reduction in distal N-terminus/C-terminus interactions in the efflux-deficient K3/5A mutant, and an increase of these interactions in the efflux-promoting S/D construct. An involvement of the distal N-terminus/C-terminus interactions in facilitating efflux is also consistent with the observation that palmitoylation of the C-terminus reduces efflux, presumably by limiting interaction with the C-terminus. This limitation would reduce N-terminus phosphorylation [52, 53] by the Ca2+/calmodulin-dependent protein kinase II (CaMKII) that is proposed to attach to the distal C-terminus of hDAT in order to phosphorylate the serine residues in the distal N-terminus for AMPH-induced efflux [25].
Conclusions
The molecular mechanisms emerging from the studies presented here offer detailed quantitative explanations for the experimental findings about the modulation of transporter function by the N-terminus of DAT, SERT, and other eukaryotic transporters. These molecular mechanisms connect the observed elongation of N- and C-termini in the evolution from bacterial homologs, with the appearance of new functional properties (e.g., efflux) that are not shared by the bacterial homologs which lack the long N-terminus but share the overall molecular architecture (termed the “LeuT-fold” [37]). They are clearly enabled by specific roles of either the proximal or distal segments, or both working in concert. Notably, the specific (different) involvements of proximal and distal segments — such as the role of the proximal segment in sustaining transport in PIP2-depleted membranes and that of the distal segment in modulating efflux — may represent an evolutionary adaptation required for the function of eukaryotic transporters expressed in various cell types of the same organism, which can differ in the lipid composition and protein complement of their membrane environment.
Methods
System preparation
The molecular model of full-length wild-type hDAT used in this study is the same as that described and investigated earlier [32]. The R51W and K3A + K5A constructs were prepared by introducing the mutations in the wild-type hDAT model using the VMD mutator plugin [54]. To build the S/D mutant (simultaneous mutations of S2, S4, S7, S12, and S13 residues to Asp), we combined, using Modeller version 9v1 [55], the 57–620 residue stretch from the wild-type hDAT structure with the structural model of the 1–57 S/D segment elaborated and described previously [20].
The full-length models of the hDAT constructs R51W, K3A + K5A (heretofore referred to as “K3/5A”), and S/D were inserted into the same pre-equilibrated compositionally asymmetric bilayer membrane used for MD simulations of the wild-type hDAT [32]. This lipid bilayer was designed to mimic a neuronal cell plasma membrane and contains 5% PIP2 lipid on the intracellular leaflet of the bilayer (see Additional file 2: Table S1 for the membrane lipid composition). For the simulations of the wild-type hDAT in the PIP2-depleted membrane environment, as done previously [29], all the PIP2 lipids in the bilayer were changed to 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) lipids, the major component of the intracellular leaflet of our model bilayer. All the hDAT-membrane systems were solvated in a 150 mM K+Cl− TIP3P water solution with ions added for neutrality, resulting in a final atom count of ~ 150,000.
Molecular dynamics simulations
All-atom MD simulations were carried out using the same scheme as described earlier for the wild-type hDAT in PIP2-enriched membranes [32]. Briefly, using NAMD software version 2.10 [56], the systems first were equilibrated following the same multi-step equilibration protocol used previously [32] during which the backbone of the protein was first fixed, then harmonically restrained, and finally released. After this equilibration phase, the velocities of all the atoms in the system were reset (at T = 310 K using random number seed), and 50 independent ~ 1-μs-long unbiased MD simulations were carried out using the latest version of the ACEMD software [57], resulting in a cumulative MD simulation time of ~ 50 μs per system. These production simulations were performed under NVT ensemble and with all the default run parameters validated by the ACEMD developers (https://www.acellera.com/) and in a large number of published applications (e.g., see https://www.acellera.com). The run parameters (4-fs time step with hydrogen mass repartitioning; particle mesh Ewald (PME) for electrostatics; switched Lennard-Jones interactions with a cutoff of 9 Ǻ, and switching distance set to 7.5 Ǻ) have been shown to reliably reproduce known values for free energy of protein folding and a variety of properties of lipid membranes [58, 59]. In addition, ensemble MD simulations with ACEMD have been generally used to generate large data sets of trajectories for quantitative analysis of kinetics of ligand-induced conformational transitions in G protein-coupled receptors (GPCRs) [60], of protein-protein association/dissociation processes [61], of phospholipid scrambling processes mediated by the GPCR opsin [62], as well as for identifying pathways for spontaneous cholesterol movement in adenosine A2A GPCR [63].
Calculation of the interaction strengths
To obtain a measure of interaction between the N-terminus and other intracellular regions of hDAT, we counted the number of interactions between charged residues from the N-terminus and the intracellular loop regions. The number of interactions was quantified for a cutoff distance of 7 Å between interacting residue pairs from head group atoms of the N-terminus and of intracellular domain residues (using Nξ for Lys, Cξ for Arg, Cδ for Glu, and Cγ for Asp). The residues included in the calculations are listed in Additional file 2: Table S2. All 50 trajectories for each construct were used for these calculations, with the first 500 ns of each trajectory considered an equilibration phase so that only the 500–940 ns time interval from each trajectory was used. This ensured that the total number of frames used for the analysis was the same for each construct. The total number of interactions was then divided by the total number of trajectories (i.e., 50) to obtain the average “interaction strength” represented in Fig. 2. Error bars were calculated by taking the standard deviation of “interaction strength” in all 50 trajectories.
Markov state-based quantitative kinetic model construction
We used the Markov state model (MSM) approach to analyze the trajectories in the absence of PIP2 lipids and build quantitative kinetic models of sodium release from the Na2 site for comparison with the kinetics and pathways calculated in the presence of PIP2 in Ref. [32]. Therefore, we have followed the same protocol here as described in detail in Ref. [32]. Such quantitative kinetic models provided by MSMs [64,65,66] have been widely applied in protein folding studies [67, 68], and MSM-based kinetic model predictions have been validated experimentally [69, 70]. We and others have used MSMs combined with reactive flux analysis, such as transition path theory (TPT) analysis, to obtain key mechanistic insights into functions of membrane proteins [32, 71, 72]. The three main components for achieving quantitative MSM-based kinetic models are briefly reviewed below.
-
1.
Dimensionality reduction using tICA. Reducing the dimensionality of a system as large and complex as the membrane-immersed hDAT is necessary in order to construct an interpretable kinetic model. A natural choice of suitable reaction coordinates is those that can project the conformational space of the system along its slowest reaction coordinate, as this reaction coordinate will capture most of the conformational heterogeneity during the time course of the simulation [73]. The time-structure-based independent component analysis (tICA) method was developed recently for this purpose of capturing the slowest reaction coordinate of a system [74,75,76]. Briefly, the tICA method involves a transformation that utilizes two matrices constructed from the trajectory data: the covariance matrix C, and a time-lagged covariance matrix C TL . The slowest reaction coordinates of a system are then defined by eigenvectors of the generalized eigenvalue problem C TL V = CVΛ, where Λ and V are the eigenvalue and eigenvector matrices, respectively. The eigenvectors corresponding to the largest eigenvalues identify the slowest reaction coordinates. Here we used a lag time of 16 ns to construct the time-lagged covariance matrix C TL , and the tICA parameters were chosen as before for the hDAT molecular system [32] to measure (a) the dynamics of the Na+ ion from the Na2 site, termed Na+/Na2, and (b) the dynamics of intracellular gates formed between residues R60, D436, R445, and E428 (Additional file 2: Table S3).
-
2.
Markov model construction. The conformational transitions of biomolecular systems where the time between transitions is long enough can be modeled as Markov chains [64] in which transitions among states depend only on the current state of the system (i.e., Markovian behavior). Such Markov models provide powerful tools for outcome prediction by enabling the extraction of long time-scale information from multiples of short time-scale events.
Two components needed for the construction of such a Markov model are an ensemble of microstates of the system, and of the transitions among these microstates [77]. Microstates are defined by clustering the conformational space of the system into several basins using automated clustering algorithms like K-means or K-centers, and this technique is most practical if performed in a dimensionality-reduced space such as the one obtained from the tICA transformation. The transitions among the microstates are calculated for a particular time interval between each of the transitions (called the lag time) and stored in the transition count matrix. By row-normalizing the transition count matrix one obtains the transition probability matrix (TPM). To validate Markovian behavior, the TPMs are constructed for multiple lag times and the relaxation timescales of the system are extracted by using the relation:
$$ {\tau}_i=-\frac{\tau^{\hbox{'}}}{\ln {\lambda}_i} $$where τ’ is the lag time used for building the TPM, λi is the ith eigenvalue of the transition probability matrix, and the resulting τi is called the implied timescale corresponding to the ith relaxation mode of the system. The system is considered to be Markovian if the behavior is such that τi is independent of τ’; the corresponding TPM is a Markovian TPM that contains all the information about the thermodynamics and kinetics of the system. Thermodynamic information is stored in the first eigenvector (which has an eigenvalue of 1). Information about kinetics is stored in the subsequent (second, third, etc.) eigenvectors, with the second eigenvector (corresponding to the second largest eigenvalue) capturing the slowest dynamics of the system, the third eigenvector capturing the second slowest dynamics, and so on.
Following the same protocols as described in detail in Ref. [32] for the construction of the Markov models, we discretized the reduced conformational space generated by the first two tICA reaction coordinates into 100 microstates (Additional file 1: Figure S11) using the K-means clustering algorithm implemented in the MSMBuilder3 software [78]. TPMs were constructed at several different lag times to obtain the implied time-scale plots shown in Additional file 1: Figure S10, so that the one in which Markovian behavior is exhibited can be identified and chosen for further analysis.
-
3.
Transition path theory analysis. In addition to the thermodynamics and kinetics information it contains, the Markovian TPM also contains mechanistic information for a dynamic system. An established means of revealing such mechanistic information inherent in the TPM is the TPT analysis that identifies the most probable flux pathways of the system [79]. TPT provides such pathways by constructing a flux matrix from the Markovian TPM. This matrix conversion has been documented in detail [79, 80], and its implementation is discussed in our previous publication [32]. Although directly applicable to MSM in the microstate space (on the order of hundreds to thousands of states), TPT analysis is usually done on a macrostate MSM (on the order of tens of states) for a better visualization of flux pathways. Here we transitioned from the microstate MSM to the macrostate MSM by using the robust Perron cluster analysis (PCCA+) algorithm [81] that lumps microstates into macrostates using the sign structure of the MSM eigenvectors (this assumes that microstates with the same signs, positive or negative, in the MSM eigenvectors, will have similar kinetics [82]). Using the PCCA+ algorithm we obtained 15 macrostates, and by applying the TPT analysis to these macrostates we obtained the most probable flux pathways for the system.
Abbreviations
- DAT:
-
Dopamine transporter
- hDAT:
-
Human dopamine transporter
- MSM:
-
Markov state model
- NSS:
-
Neurotransmitter:sodium symporter
- SERT:
-
Serotonin transporter
- SLC6:
-
Solute carrier 6
- tICA:
-
Time-structure-based independent component analysis
- TM:
-
Transmembrane
- TPM:
-
Transition probability matrix
- TPT:
-
Transition path theory
References
Broer S, Gether U. The solute carrier 6 family of transporters. Br J Pharmacol. 2012;167:256–78.
Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–33.
Ng J, Papandreou A, Heales SJ, Kurian MA. Monoamine neurotransmitter disorders—clinical advances and future perspectives. Nat Rev Neurol. 2015;11:567–84.
Vaughan RA, Foster JD. Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol Sci. 2013;34:489–96.
Ritz MC, Lamb R, Kuhar M. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23.
Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–49.
Shekar A, Aguilar JI, Galli G, Cozzi NV, Brandt SD, Ruoho AE, Baumann MH, Matthies HJ, Galli A. Atypical dopamine efflux caused by 3, 4-methylenedioxypyrovalerone (MDPV) via the human dopamine transporter. J Chem Neuroanat. 2017;83:69–74.
Bowton E, Saunders C, Erreger K, Sakrikar D, Matthies HJ, Sen N, Jessen T, Colbran RJ, Caron MG, Javitch JA. Dysregulation of dopamine transporters via dopamine D2 autoreceptors triggers anomalous dopamine efflux associated with attention-deficit hyperactivity disorder. J Neurosci. 2010;30:6048–57.
Bowton E, Saunders C, Reddy IA, Campbell NG, Hamilton PJ, Henry LK, Coon H, Sakrikar D, Veenstra-VanderWeele JM, Blakely RD, Sutcliffe J, Matthies HJ, Erreger K, Galli A. SLC6A3 coding variant Ala559Val found in two autism probands alters dopamine transporter function and trafficking. Transl Psychiatry. 2014;4:e464.
Hamilton PJ, Campbell NG, Sharma S, Erreger K, Herborg Hansen F, Saunders C, Belovich AN, Sahai MA, Cook EH, Gether U, McHaourab HS, Matthies HJ, Sutcliffe JS, Galli A. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry. 2013;18:1315–23.
Hansen FH, Skjorringe T, Yasmeen S, Arends NV, Sahai MA, Erreger K, Andreassen TF, Holy M, Hamilton PJ, Neergheen V, Karlsborg M, Newman AH, Pope S, Heales SJ, Friberg L, Law I, Pinborg LH, Sitte HH, Loland C, Shi L, Weinstein H, Galli A, Hjermind LE, Moller LB, Gether U. Missense dopamine transporter mutations associate with adult parkinsonism and ADHD. J Clin Invest. 2014;124:3107–20.
Mazei-Robison MS, Bowton E, Holy M, Schmudermaier M, Freissmuth M, Sitte HH, Galli A, Blakely RD. Anomalous dopamine release associated with a human dopamine transporter coding variant. J Neurosci. 2008;28:7040–6.
Mergy MA, Gowrishankar R, Gresch PJ, Gantz SC, Williams J, Davis GL, Wheeler CA, Stanwood GD, Hahn MK, Blakely RD. The rare DAT coding variant Val559 perturbs DA neuron function, changes behavior, and alters in vivo responses to psychostimulants. Proc Natl Acad Sci. 2014;111:E4779–88.
Ng J, Zhen J, Meyer E, Erreger K, Li Y, Kakar N, Ahmad J, Thiele H, Kubisch C, Rider NL. Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain. 2014;137:1107–19.
Sakrikar D, Mazei-Robison MS, Mergy MA, Richtand NW, Han Q, Hamilton PJ, Bowton E, Galli A, Veenstra-Vanderweele J, Gill M, Blakely RD. Attention deficit/hyperactivity disorder-derived coding variation in the dopamine transporter disrupts microdomain targeting and trafficking regulation. J Neurosci. 2012;32:5385–97.
Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl–-dependent neurotransmitter transporters. Nature. 2005;437:215–23.
Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90.
Malinauskaite L, Quick M, Reinhard L, Lyons JA, Yano H, Javitch JA, Nissen P. A mechanism for intracellular release of Na+ by neurotransmitter/sodium symporters. Nat Struct Mol Biol. 2014;21:1006–12.
Kantcheva AK, Quick M, Shi L, Winther A-ML, Stolzenberg S, Weinstein H, Javitch JA, Nissen P. Chloride binding site of neurotransmitter sodium symporters. Proc Natl Acad Sci. 2013;110:8489–94.
Khelashvili G, Doktorova M, Sahai MA, Johner N, Shi L, Weinstein H. Computational modeling of the N-terminus of the human dopamine transporter and its interaction with PIP2-containing membranes. Proteins. 2015;83:952–69.
Fenollar-Ferrer C, Stockner T, Schwarz TC, Pal A, Gotovina J, Hofmaier T, Jayaraman K, Adhikary S, Kudlacek O, Mehdipour AR. Structure and regulatory interactions of the cytoplasmic terminal domains of serotonin transporter. Biochemistry. 2014;53:5444–60.
Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:E78.
Moritz AE, Rastedt DE, Stanislowski DJ, Shetty M, Smith MA, Vaughan RA, Foster JD. Reciprocal phosphorylation and palmitoylation control dopamine transporter kinetics. J Biol Chem. 2015;290:29095–105.
Binda F, Dipace C, Bowton E, Robertson SD, Lute BJ, Fog JU, Zhang M, Sen N, Colbran RJ, Gnegy ME. Syntaxin 1A interaction with the dopamine transporter promotes amphetamine-induced dopamine efflux. Mol Pharmacol. 2008;74:1101–8.
Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N, Nikandrova Y, Bowton E, McMahon DG, Colbran RJ. Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron. 2006;51:417–29.
Tavoulari S, Margheritis E, Nagarajan A, DeWitt DC, Zhang Y-W, Rosado E, Ravera S, Rhoades E, Forrest LR, Rudnick G. Two Na+ sites control conformational change in a neurotransmitter transporter homolog. J Biol Chem. 2016;291:1456–71.
LeVine MV, Cuendet MA, Razavi AM, Khelashvili G, Weinstein H. Thermodynamic coupling function analysis of allosteric mechanisms in the human dopamine transporter. Biophys J. 2018;114:10–4.
LeVine MV, Cuendet MA, Khelashvili G, Weinstein H. Allosteric mechanisms of molecular machines at the membrane: transport by sodium-coupled symporters. Chem Rev. 2016;116:6552–87.
Khelashvili G, Stanley N, Sahai MA, Medina J, LeVine MV, Shi L, De Fabritiis G, Weinstein H. Spontaneous inward opening of the dopamine transporter is triggered by PIP2-regulated dynamics of the N-terminus. ACS Chem Nerosci. 2015;6:1825–37.
Cheng MH, Bahar I. Molecular mechanism of dopamine transport by human dopamine transporter. Structure. 2015;23:2171–81.
Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter—inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30:667–77.
Razavi AM, Khelashvili G, Weinstein H. A Markov state-based quantitative kinetic model of sodium release from the dopamine transporter. Sci Rep. 2017;7:40076.
Cartier E, Hamilton P, Belovich AN, Shekar A, Campbell NG, Saunders C, Andreassen TF, Gether U, Veenstra-Vanderweele J, Sutcliffe JS, Ulery-Reynolds PG, Erreger K, Matthies HJ, Galli A. Rare autism-associated variants implicate syntaxin 1 (STX1 R26Q) phosphorylation and the dopamine transporter (hDAT R51W) in dopamine neurotransmission and behaviors. EBioMedicine. 2015;2:135–46.
Hamilton PJ, Belovich AN, Khelashvili G, Saunders C, Erreger K, Javitch JA, Sitte HH, Weinstein H, Matthies HJ, Galli A. PIP2 regulates psychostimulant behaviors through its interaction with a membrane protein. Nat Chem Biol. 2014;10:582–9.
Kern C, Erdem FA, El-Kasaby A, Sandtner W, Freissmuth M, Sucic S. The N terminus specifies the switch between transport modes of the human serotonin transporter. J Biol Chem. 2017;292:3603–13.
Anderluh A, Hofmaier T, Klotzsch E, Kudlacek O, Stockner T, Sitte HH, Schütz GJ. Direct PIP2 binding mediates stable oligomer formation of the serotonin transporter. Nat Commun. 2017;8:14089.
Penmatsa A, Gouaux E. How LeuT shapes our understanding of the mechanisms of sodium-coupled neurotransmitter transporters. J Physiol. 2013;592:863–9.
Gether U, Andersen PH, Larsson OM, Schousboe A. Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci. 2006;27:375–83.
Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Mol Pharmacol. 2006;70:1630–42.
Li J, Wen P-C, Moradi M, Tajkhorshid E. Computational characterization of structural dynamics underlying function in active membrane transporters. Curr Opin Struct Biol. 2015;31:96–105.
Buchmayer F, Schicker K, Steinkellner T, Geier P, Stübiger G, Hamilton PJ, Jurik A, Stockner T, Yang J-W, Montgomery T. Amphetamine actions at the serotonin transporter rely on the availability of phosphatidylinositol-4, 5-bisphosphate. Proc Natl Acad Sci. 2013;110:11642–7.
Sweeney CG, Tremblay BP, Stockner T, Sitte HH, Melikian HE. Dopamine transporter amino and carboxyl termini synergistically contribute to substrate and inhibitor affinities. J Biol Chem. 2017;292:1302–9.
Moritz AE, Foster JD, Gorentla BK, Mazei-Robison MS, Yang J-W, Sitte HH, Blakely RD, Vaughan RA. Phosphorylation of dopamine transporter serine 7 modulates cocaine analog binding. J Biol Chem. 2013;288:20–32.
Foster JD, Yang J-W, Moritz AE, ChallaSivaKanaka S, Smith MA, Holy M, Wilebski K, Sitte HH, Vaughan RA. Dopamine transporter phosphorylation site threonine 53 regulates substrate reuptake and amphetamine-stimulated efflux. J Biol Chem. 2012;287:29702–12.
Cremona ML, Matthies HJ, Pau K, Bowton E, Speed N, Lute BJ, Anderson M, Sen N, Robertson SD, Vaughan RA. Flotillin-1 is essential for PKC-triggered endocytosis and membrane microdomain localization of DAT. Nat Neurosci. 2011;14:469–77.
Sucic S, Dallinger S, Zdrazil B, Weissensteiner R, Jørgensen TN, Holy M, Kudlacek O, Seidel S, Cha JH, Gether U. The N terminus of monoamine transporters is a lever required for the action of amphetamines. J Biol Chem. 2010;285:10924–38.
Li J, Shaikh SA, Enkavi G, Wen P-C, Huang Z, Tajkhorshid E. Transient formation of water-conducting states in membrane transporters. Proc Natl Acad Sci. 2013;110:7696–701.
Shaikh SA, Tajkhorshid E. Modeling and dynamics of the inward-facing state of a Na+/Cl− dependent neurotransmitter transporter homologue. PLoS Comput Biol. 2010;6:e1000905.
Zomot E, Gur M, Bahar I. Microseconds simulations reveal a new sodium-binding site and the mechanism of sodium-coupled substrate uptake by LeuT. J Biol Chem. 2015;290:544–55.
Cheng MH, Bahar I. Complete mapping of substrate translocation highlights the role of LeuT N-terminal segment in regulating transport cycle. PLoS Comput Biol. 2014;10:e1003879.
Khelashvili G, Weinstein H. Functional mechanisms of neurotransmitter transporters regulated by lipid-protein interactions of their terminal loops. Biochim Biophys Acta. 2015;1848:1765–74.
Foster JD, Vaughan RA. Phosphorylation mechanisms in dopamine transporter regulation. J Chem Neuroanat. 2016;83:10–8.
Rastedt D, Foster J, Vaughan R. Dopamine transporter expression and transport capacity is regulated by palmitoylation. FASEB J. 2015;29:768.764.
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8.
Webb B, Sali A. Protein structure modeling with MODELLER. In: Kihara D, editor. Protein struct prediction. Methods in molecular biology (methods and protocols), vol. 1137. New York: Humana Press; 2014. p. 1–15.
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802.
Harvey MJ, Giupponi G, Fabritiis GD. ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput. 2009;5:1632–9.
Dainese E, De Fabritiis G, Sabatucci A, Oddi S, Angelucci CB, Di Pancrazio C, Giorgino T, Stanley N, Del Carlo M, Cravatt BF. Membrane lipids are key modulators of the endocannabinoid-hydrolase FAAH. Biochem J. 2014;457:463–72.
Piana S, Lindorff-Larsen K, Dirks RM, Salmon JK, Dror RO, Shaw DE. Evaluating the effects of cutoffs and treatment of long-range electrostatics in protein folding simulations. PLoS One. 2012;7:e39918.
Kapoor A, Martinez-Rosell G, Provasi D, Fabritiis G, Filizola M. Dynamic and kinetic elements of μ-opioid receptor functional selectivity. Sci Rep. 2017;7:11255.
Plattner N, Doerr S, De Fabritiis G, Noé F. Complete protein–protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling. Nat Chem. 2017;9:1005–11.
Morra G, Razavi AM, Pandey K, Weinstein H, Menon AK, Khelashvili G. Mechanisms of lipid scrambling by the G protein-coupled receptor opsin. Structure. 2017;26:1–12.
Guixà-González R, Albasanz JL, Rodriguez-Espigares I, Pastor M, Sanz F, Martí-Solano M, Manna M, Martinez-Seara H, Hildebrand PW, Martín M. Membrane cholesterol access into a G-protein-coupled receptor. Nat Commun. 2017;8:14505.
Chodera JD, Swope WC, Pitera JW, Dill KA. Long-time protein folding dynamics from short-time molecular dynamics simulations. Multiscale Model Simul. 2006;5:1214–26.
Prinz J-H, Wu H, Sarich M, Keller B, Senne M, Held M, Chodera JD, Schütte C, Noé F. Markov models of molecular kinetics: generation and validation. J Chem Phys. 2011;134:174105.
Pande VS, Beauchamp K, Bowman GR. Everything you wanted to know about Markov State Models but were afraid to ask. Methods. 2010;52:99–105.
Voelz VA, Bowman GR, Beauchamp K, Pande VS. Molecular simulation of ab initio protein folding for a millisecond folder NTL9 (1−39). J Am Chem Soc. 2010;132:1526–8.
Beauchamp KA, McGibbon R, Lin YS, Pande VS. Simple few-state models reveal hidden complexity in protein folding. Proc Natl Acad Sci U S A. 2012;109:17807–13.
Lapidus LJ, Acharya S, Schwantes CR, Wu L, Shukla D, King M, DeCamp SJ, Pande VS. Complex pathways in folding of protein G explored by simulation and experiment. Biophys J. 2014;107:947–55.
Voelz VA, Jäger M, Yao S, Chen Y, Zhu L, Waldauer SA, Bowman GR, Friedrichs M, Bakajin O, Lapidus LJ. Slow unfolded-state structuring in Acyl-CoA binding protein folding revealed by simulation and experiment. J Am Chem Soc. 2012;134:12565–77.
Kohlhoff KJ, Shukla D, Lawrenz M, Bowman GR, Konerding DE, Belov D, Altman RB, Pande VS. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nat Chem. 2014;6:15–21.
Adelman JL, Ghezzi C, Bisignano P, Loo DD, Choe S, Abramson J, Rosenberg JM, Wright EM, Grabe M. Stochastic steps in secondary active sugar transport. Proc Natl Acad Sci. 2016;113:E3960–6.
Perez-Hernandez G, Paul F, Giorgino T, De Fabritiis G, Noe F. Identification of slow molecular order parameters for Markov model construction. J Chem Phys. 2013;139:015102.
Schwantes CR, Pande VS. Improvements in Markov State Model construction reveal many non-native interactions in the folding of NTL9. J Chem Theory Comput. 2013;9:2000–9.
Oja E, Hyvarinen A. Independent component analysis: algorithms and applications. Neural Netw. 2000;13:411–30.
Naritomi Y, Fuchigami S. Slow dynamics in protein fluctuations revealed by time-structure based independent component analysis: the case of domain motions. J Chem Phys. 2011;134:065101.
Beauchamp KA, Bowman GR, Lane TJ, Maibaum L, Haque IS, Pande VS. MSMBuilder2: modeling conformational ynamics at the picosecond to millisecond scale. J Chem Theory Comput. 2011;7:3412–9.
Harrigan MP, Sultan MM, Hernández CX, Husic BE, Eastman P, Schwantes CR, Beauchamp KA, McGibbon RT, Pande VS. MSMBuilder: statistical models for biomolecular dynamics. Biophys J. 2017;112:10–5.
Berezhkovskii A, Hummer G, Szabo A. Reactive flux and folding pathways in network models of coarse-grained protein dynamics. J Chem Phys. 2009;130:205102.
Kube S, Weber M. A coarse graining method for the identification of transition rates between molecular conformations. J Chem Phys. 2007;126:024103.
Deuflhard P, Weber M. Robust Perron cluster analysis in conformation dynamics. Linear Algebra Appl. 2005;398:161–84.
Chodera JD, Noé F. Markov state models of biomolecular conformational dynamics. Curr Opin Struct Biol. 2014;25:135–44.
McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, Schwantes CR, Wang L-P, Lane TJ, Pande VS. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J. 2015;109:1528–32.
Acknowledgements
The following computational resources are gratefully acknowledged: resources of the Oak Ridge Leadership Computing Facility (ALCC allocation BIP109) at the Oak Ridge National Laboratory, which is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC05-00OR22725; an allocation at the National Energy Research Scientific Computing Center (NERSC, repository m1710) supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02- 05CH11231; and the computational resources of the David A. Cofrin Center for Biomedical Information in the HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine at Weill Cornell Medical College.
Funding
The work was supported by National Institutes of Health (NIH) Grants P01 DA012408, R01 DA041510, and in part U54 GM087519.
Availability of data and materials
All simulation trajectories are available upon request.
Author information
Authors and Affiliations
Contributions
All authors contributed to conceiving the project and experimental design, AMR did the analysis and carried out the MSM construction, GK carried out the MD simulations, and all authors participated in the interpretation of results and in writing the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Supplementary Figures. Figure S1. Na2 distance time evolution for no-PIP2 system. Figure S2. Na2 distance time evolution for R51W system. Figure S3. Na2 distance time evolution for S/D system. Figure S4. Na2 distance time evolution for K3A/K5A system. Figure S5. Intracellular gating residues distances for S/D system. Figure S6. Intracellular gating residues distances for R51W system. Figure S7. Intracellular gating residues distances for no-PIP2 system. Figure S8. Frequencies of intracellular gating residues interactions. Figure S9. Distribution of the R445–E428 distance in different constructs of hDAT. Figure S10. MSM implied-timescales plots. Figure S11. MSM 1st relaxation mode for no-PIP2 system. Figure S12. Localization density of the N-terminus near the membrane. Figure S13. MSM macrostates for the no-PIP2 systems. (PDF 9.96 mb)
Additional file 2:
Supplementary Tables. Table S1. Lipid composition of the model membrane. Table S2. Residues included in the calculation of interaction strength between N-terminus and intracellular regions. Table S3. Parameters used for dimensionality reduction and tICA construction. Table S4. Correlation values for water coordination number. Table S5. MSM predicted sodium release pathways. (PDF 169 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Razavi, A.M., Khelashvili, G. & Weinstein, H. How structural elements evolving from bacterial to human SLC6 transporters enabled new functional properties. BMC Biol 16, 31 (2018). https://doi.org/10.1186/s12915-018-0495-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-018-0495-6