
Abstract
Background
Pain is a common and distressing symptom in people with cancer, but is under-recognised and under-treated. Australian guidelines for ‘Cancer Pain Management in Adults’ are available on the Cancer Council Australia Cancer Guideline Wiki. This study aims to evaluate the effectiveness and cost-effectiveness of a suite of guideline implementation strategies for improving pain outcomes in adults with cancer in oncology and palliative care outpatient settings.
Methods
The study will use a stepped-wedge cluster randomised controlled design, with oncology and palliative care outpatient services as the clusters. Patients will be eligible if they are adults with cancer and pain presenting to participating services during the study period. During an initial control arm, services will routinely screen patients for average and worst pain over the past 24 h using a 0–10 numerical rating scale (NRS) and have unfettered access to online guidelines. During the intervention arm, staff at each service will be encouraged to use: 1) a patient education booklet and self-management resource; 2) an online spaced learning cancer pain education module for clinicians from different disciplines; and 3) audit and feedback of service performance on key indices of cancer pain screening, assessment and management. Service-based clinical change champions will lead implementation of these strategies.
The trial’s primary outcome will be the probability that patients initially screened as having moderate-severe (≥5/10 NRS) worst pain experience a clinically important improvement one week later, defined as ≥ 30% reduction. Secondary outcomes will include patient empowerment and quality of life, carer experience, and cost-effectiveness. For the main analysis, linear mixed models will be used, accounting for clustering and the longitudinal design. Eighty-two patients per service at six services (N = 492) will provide > 90% power. A qualitative sub-study and analyses of structural and process factors will explore opportunities for further refinement and tailoring of the intervention.
Discussion
This pragmatic trial will inform implementation of guidelines across a range of oncology and palliative care outpatient service contexts. If found effective, the implementation strategies will be made freely available on the Wiki alongside the guidelines.
Trial registration
Registered 23/01/2015 on the Australian New Zealand Clinical Trials Registry (ACTRN12615000064505).
Similar content being viewed by others


Background
Despite of decades of research and practice improvement initiatives, 39–66% of people with cancer experience pain, and up to 40% of people with pain receive inadequate analgesia [1, 2]. Under-treated pain not only reduces patient quality of life (QOL) [3] but also increases health service use and costs [4]. Barriers to pain assessment and management occur at the levels of the patient (e.g. reluctance to report pain, misconceptions regarding opioids), clinician (e.g. lack of time and expertise), service (e.g. inadequate process for screening) and healthcare system (e.g. lack of coordination) [5,6,7,8,9,10]. Routine screening and implementation of evidence-based guidelines can improve quality of care and outcomes for cancer pain [11,12,13]. However, clinicians are unlikely to use symptom screening results or guidelines unless they are motivated and supported by focused strategies [14, 15].
The current authors have undertaken a program of work to develop and implement Australian guidelines for ‘Cancer Pain Management in Adults’, which are now available online via the Cancer Council Australia Cancer Guidelines Wiki [16, 17]. This program was structured in accordance with the UK Medical Research Council’s (MRC) Framework for complex interventions, defined as those with multiple interacting components that need tailoring to local healthcare settings and other contexts [18, 19]. The MRC recommends that complex intervention programs consist of four phases: development, feasibility and piloting, evaluation, and implementation. The development phase of our program involved a national survey of current practice [20,21,22], two systematic reviews [5, 23] and consideration of the wider literature by an Organising Committee within a theoretical framework called the Behaviour Change Wheel [24]. This process identified three strategies showing promise for improving care processes and outcomes for cancer pain: patient training, clinician education, and audit and feedback. These strategies are discussed in detail in the Methods section of the current paper. Our piloting phase tested the feasibility of screening for pain and auditing pain assessment and management in adults attending palliative care and oncology services [25]. The current paper focuses on the protocol for the evaluation phase, which aims to test the effectiveness and cost-effectiveness of all three guideline implementation strategies combined for improving pain outcomes in adults with cancer attending oncology and palliative care outpatient services.
Methods/design
Aims
Primary aim
The study’s primary aim is to evaluate capacity for a suite of three guideline implementation strategies to increase the probability that patients initially screened as having moderate-severe (≥5) worst pain on a 0–10 numerical rating scale (NRS) will experience a clinically important improvement of ≥30% one week later.
Secondary aims
Secondary aims are:
-
1.
to evaluate capacity of the three guideline implementation strategies versus a control arm to:
-
i.
reduce mean worst and average pain severity across all patients screened as having clinically relevant (≥2 NRS) or moderate-severe (≥5 NRS) worst pain from time of screening to one, two and four week later by half a standard deviation (0.5 SD);
-
ii.
demonstrate a between-arm difference in patient empowerment in patients screened as having clinically relevant (≥2 NRS) worst pain, as measured by 0.5 SD difference on the Health Education Impact Questionnaire (heiQ) (41) at one, two and four weeks post-screening;
-
iii.
demonstrate a between-arm difference in the experiences of unpaid carers of participating patients, as measured by a 0.5 SD difference on the Carer Experience Scale (CES) [26] at two and four weeks post-screening;
-
iv.
demonstrate a between-arm difference on mean patient quality of life (QOL), as measured by a difference of 0.5 SD on the European Organisation for Research and Treatment of Cancer Palliative Care (EORTC QLQ C15-PAL) [27] at two and four weeks post-screening;
-
2.
to evaluate the cost-effectiveness of the three guideline implementation strategies based on incremental cost per additional responder on the primary outcome;
-
3.
to inform refinement and tailoring of the implementation strategies for different service settings.
Design and setting
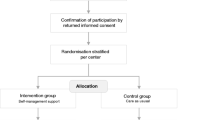
The study will use a stepped-wedge cluster randomised controlled trial (RCT) design, with Australian oncology and palliative care services as the units of randomisation. In this design, randomisation concerns the sequence in which services transition between experimental conditions rather than the condition to which they are allocated (see Fig. 1). Each participating patient and carer will contribute data to only one arm, not both.

Stepped wedge design with staggered introduction of training/intervention in 8 services
While needing a longer timeline than a parallel cluster design, a stepped-wedge design has the following advantages for translational research: 1) the design controls for between-service variation in baseline practice; 2) statistical power is boosted by the opportunity to assess intervention effects in a pre/post comparison across services; and 3) assessment of longitudinal effects (i.e. sustainability) can occur in services that transition earlier [28].
Ethics approval was granted by the South Western Sydney Local Health District Human Research Ethics Committee (HREC). Protocol modifications will be submitted to the HREC for approval, and modifications made to the trial registry information as needed.
This protocol complies with reporting requirements outlined in the cluster RCT extension of the Consolidated Standards of Reporting Trials (CONSORT) [29] and Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) [30].
Participants
Services
Services will be selected to inform understanding of intervention effectiveness across services of differing geographic locations (e.g. state/territories; metropolitan, regional, rural) and configurations of oncology and palliative care. Study sites are included in the trial’s entry on the Australian New Zealand Clinical Trials Registry.
Patients
There will be two patient participant populations for this study. The first will contribute to primary outcome data, and the second to secondary outcomes. Because the study is funded by the National Breast Cancer Foundation (NBCF) of Australia, sampling will aim to recruit ≥60% patients with breast cancer. It is anticipated that breast cancer’s prevalence and this population’s proven willingness to participate in clinical trials [31] will make over-sampling unnecessary.
Participant eligibility criteria will follow recommendations from the Methods for Researching End of Life Care (MORECare) initiative [32] by targeting people most likely to benefit from the intervention. Inclusion criteria for patients to contribute to primary outcome will be: 1) attending a participating service as an outpatient during the study period; 2) having a diagnosis of advanced cancer of any type; 3) ability to self-complete a 0–10 NRS for severity of worst and average pain in English or one of five widely-spoken languages most commonly associated with poor English proficiency in Australia (Chinese, Italian, Greek, Vietnamese and Arabic) [33]; 4) choosing not to opt-out of being contacted one week later to complete the NRS and giving verbal informed consent to participate when contacted; and 5) a score of ≥5 on the NRS for worst pain. Inclusion criteria for patients contributing to secondary outcomes will be the same as criteria one, two, three and four above, as well as: 5) a score of ≥2 on the NRS for worst pain; and 6) spoken English proficiency sufficient to complete secondary outcome measures.
Patients will be excluded if they have already participated in the study either at another service taking part in the trial or at the same service when it was in the control arm, or are documented as having cognitive impairment that would preclude capacity to give informed consent.
Potentially eligible patients will be identified by the research team via a review of service pain screening data (see below), which will include information about their NRS score, whether they have opted out of being contacted, and their cancer status (completed by clinic staff). Patients will be telephoned at home, and the verbal informed consent procedure undertaken.
Carers
Unpaid carers will be eligible if they: 1) are identified by a patient who has given informed consent to participate in the study as providing them with substantial emotional and practical support in an unpaid capacity; 2) are willing to provide verbal informed consent to participate; and 3) have sufficient spoken English proficiency to complete a survey and/or interview.
Carers will be excluded if the patient for whom they provide care does not participate in any secondary outcome components.
Service staff
Service staff will be eligible to participate if they are: 1) employed on a permanent basis either full- or part-time at a participating service in a role that provides clinical care to patients with cancer pain or front desk administrative support; and 2) provide informed consent.
Service staff will be excluded if they are working at a participating service on a casual or agency basis.
Intervention
Each participating service will first be included in the control arm and then transition to the intervention arm, defined as follows. Due to the pragmatic nature of the trial, no limit is placed on other interventions each service can undertake during the study period.
Control arm
We will compare the intervention with usual care plus the introduction of routine screening for pain where this is not already common practice. The rationale for including a screening system in the usual care arm is that this is already routine practice in some oncology and palliative care services across Australia.
Management and clinicians at participating services will be made aware of the Australian online guidelines for ‘Cancer Pain Management in Adults’ and the study’s aim to test implementation of these. Services that are already utilising an electronic or paper-based symptom screening system will be asked to incorporate the study’s NRS for worst and average pain over the past 24 h. Where no screening system is established, services will be equipped with a paper-based system for symptom screening and providing individual patient reports for treating clinicians to refer to during consultations. A paper-based rather than electronic screening system has been chosen because a pilot study conducted by the current authors found that implementing electronic screening can be resource-intensive and logistically challenging [25]. Acknowledging that migrants with limited proficiency in their host-country language may be at special risk of poor outcomes [34], screening measures will be made available in five widely-spoken languages most commonly associated with poor English proficiency in Australia (Chinese, Italian, Greek, Vietnamese and Arabic) [33].
While access to pain screening results and online guidelines will be unfettered, services will not be given strategies to encourage or support clinicians in using these.
The control arm at each service will continue as long as necessary to recruit the target sample size.
Intervention arm
At the beginning of a training phase, two clinicians at each service will take on the role of ‘clinical change champions’ to lead local implementation of the intervention with support from the project team. Two are needed to cover any periods of absence. Selection of clinical change champions will be conducted in partnership with managers at each service. Champions can be of any discipline and role provided they meet established criteria [35].
Champions will attend training at their service aimed at tailoring the intervention to local needs and contexts. Champions will be involved in administering the audit tool and feeding back data to staff in order to identify strengths and weaknesses at their service against recommendations in the guidelines. They will also be supported to identify barriers and facilitators to cancer pain management and helped to develop solutions. Champions will be invited to attend monthly teleconferences with champions at other services to create a community of practice that will enable knowledge sharing, knowledge creation and identity building [36]. The degree to which champions take on all responsibilities or delegate these to other staff will be tailored to each service.
Also during the training phase, all clinical and reception-desk staff will be given an overview of the guidelines and implementation resources by the champions at their service with support from the project team.
During the training phase and for a period of three weeks afterwards, data will not contribute to outcome measurement to allow time for the intervention to become established as routine practice.
During the intervention phase, the process for feeding back individual patient results from screening will be monitored, with medical teams receiving a summary report of screening data for each patient prior to consultation to inform management, and a copy filed in the medical record. The process by which this happens will vary across services according to local systems.
Three implementation strategies will be used to address commonly reported barriers and facilitators in the most parsimonious way. Findings from a systematic review suggest that coverage of barriers, rather than number of strategies, may be important in successfully implementing guidelines [37]. Therefore, one strategy was chosen to intervene at each of the levels of patient, clinician and health service, with strategies designed to address barriers across more than one level wherever possible. See Table 1 for mapping of the strategies against behaviour change ‘functions’ as identified by Michie et al.’s Behaviour Change Wheel [24].
The patient-level strategy includes a written resource to help patients report and self-manage their pain, support coordination and communication between patients and clinicians, and advocate for evidence-based person-centred care [38]. This resource was designed for use in conjunction with an education booklet and DVD designed by members of the current team (ML, FB) called ‘Overcoming Cancer Pain’, published by Cancer Council Australia [39] and widely available in Australian oncology and palliative care services. The booklet includes information on pain, general advice on non-pharmacological and pharmacological management, and a pain diary template. The booklet and DVD were found to reduce pain versus usual care in a previous RCT [40]. The self-management resource includes: 1) a template for setting specific, measurable, achievable, relevant and time-bound (SMART) care goals, as well as identifying potential obstacles and ways to overcome these, and 2) an action plan detailing exacerbating and alleviating factors, current pharmacological and non-pharmacological management strategies, and contacts for support. Interviews with patients have provided preliminary support for the usefulness of the self-management resource [38, 41]. Patients and their physicians will set goals and complete the plan together to ensure shared understanding and decision-making. Patients will be encouraged to use the resource as a patient-held record that they can take to every healthcare appointment to support communication and ensure that care is coordinated and centred on their needs.
The strategy selected for the clinician level is a spaced learning training module for clinicians of different disciplines aimed at improving knowledge of guideline recommendations and applying this within the context of person-centred care. Spaced learning will be delivered online using the QStream Healthcare Solutions platform. The spaced learning format has been shown to significantly improve knowledge and retention of guideline content in RCTs [42]. This approach was chosen because it is a feasible form of education within busy clinical environments and is applicable to all members of multi-disciplinary teams. Questions testing knowledge of guideline recommendations are delivered to participants via email, which also provides immediate feedback about whether the questions have been answered correctly. Incorrect answers are resubmitted to participants at a later date and only retired when they have been answered correctly on two occasions. The use of clinical vignettes in each question ensures that learning is focused on the needs of an individual patient rather than only on biomedical aspects. Members of the current team (JP, TS) have developed and piloted a spaced learning module for cancer pain assessment in the palliative care setting, where it was associated with increased levels of nurse knowledge and documentation of pain, as well as reduced pain severity for patients [43, 44]. Further modules are being developed by the team to test and improve clinician understanding of cancer pain assessment and management.
At the service level, an audit and feedback mechanism has been chosen as the best strategy following evidence from a Cochrane systematic review that this strategy can lead to improvement in provider compliance with desired practice [45], including implementation of cancer pain guidelines [46]. The team adapted an audit tool developed by Victoria Health, which collects data on key indices of cancer pain care, including screening, assessment, regular and breakthrough analgesia, and management of adverse effects [47]. During the pilot phase of this program, the audit tool was found feasible to implement, and changes were made to improve inter-rater reliability and relevance to the current intervention [25]. In accordance with best-practice, feedback will be provided in written (e.g. newsletter) as well as verbal formats and accompanied by discussion aimed at setting and meeting SMART targets via an agreed practice improvement plan [48]. Discussions will be framed within a Plan-Do-Study-Act (PDSA) model for quality improvement, with which Australian clinicians are familiar [49].
During a sustainability phase, service clinicians who are trained in the use of the audit tool will provide the research team with de-identified audit data on pain assessment and management for patients screened as having a severe (≥7 NRS) pain score to assess whether adherence to guideline recommendations has continued.
Outcomes
The study period for each patient will be four weeks or until death, whichever is the shorter time. See Table 2 for the enrolment and assessment schedule.
Primary outcome
The primary outcome will relate to both individual patient and cluster levels and concern the probability that patients at each service initially screened as having moderate-severe (≥5 NRS) worst pain will experience a clinically important improvement one week later, defined as a ≥ 30% reduction in their original rating.
One week following positive screen is considered sufficient time to allow for comprehensive assessment and any new treatment regimens to become established. Pain over the past 24 h was chosen to balance the need to avoid recall bias with sampling a representative time window. The NRS is the optimal brief measure of pain severity based on compliance rates, responsiveness, ease of use and applicability [50]. The NRS is also the response option used by most widely used and validated multi-dimensional pain scales (e.g. Brief Pain Inventory [51]). A rating of 5 on a 0–10 NRS has become established as the threshold for moderate cancer pain [52]. Patients with pain of this magnitude or higher will form the focus of the primary endpoint because of the added urgency of reducing moderate-severe pain versus mild.
A 30% reduction in pain was selected as the minimal clinically important difference (MCID) based on recommendations for pain trials by the US Food and Drug Administration, which takes into account evidence that the number of NRS points needed for a MCID varies according to the baseline score [53].
To minimise selection bias, the study will use an opt-out procedure for patients’ permission to obtain their contact details and telephone them one week later to measure the primary outcome. Upon contact, patients will be invited to give verbal informed consent to provide primary outcome data, and will remain at liberty to refuse at this time.
For a flow diagram of recruitment of patients to the primary outcome component of this study, see Fig. 2.

Study diagram of patient recruitment and data collection for primary endpoint of worst pain over past 24 h at one week after screening with worst pain of ≥5 on a 0–10 numerical rating scale (NRS)
Secondary outcomes
Secondary outcomes will assume a MCID to be 0.5 standard deviation (SD), using an established rule of thumb for patient reported outcomes [54].
Patient pain
Secondary outcomes for pain will be at the cluster level and relate to the potential for the intervention versus control arm to result in: 1) mean reduction for worst pain severity in patients with clinically significant (≥2 NRS) and moderate-severe (≥5 NRS) worst pain from screening to one, two and four weeks later; and 2) mean reduction for worst and average pain across all screenings, to give a population measure of effect.
A waiver on patient consent has been authorised by the HREC to include de-identified pain screening data from all patients attending services during the control and intervention arms to avoid selection bias. However, informed consent will be required for participation in all other secondary outcomes, detailed as follows. The HREC has approved a verbal (rather than written) consent procedure for all outcomes except health service utilisation, for which written informed consent will be required using a standardised form developed by Medicare.
Patient quality of life
Patient QOL will be measured at weeks one, two and 4 using the EORTC QLQ C15-PAL [27], a short-form of the EORTC QLQ-C30 [55], which is among the most widely used cancer-specific QOL measures. The QLQ C15-PAL includes 15 items to reduce burden and focus on issues most relevant to people with advanced cancer. It provides scores for global QOL, physical functioning, psychological functioning, and symptoms that include fatigue, nausea and vomiting, breathlessness, insomnia, appetite loss and constipation. Importantly for this study, it also provides a score for pain interference and uses a recall period of the past week; both features will complement the two pain NRS, which focus on pain severity over the past 24 h. In this study, five additional items from the QLQ-C30 will be added to the C15-PAL at every administration to enable cost-utility evaluation using the QLQ-Utility measure [56]. These items assess physical functioning, role functioning, social functioning (two items) and diarrhea.
A MCID of 0.5 SD difference between control and intervention arms will provide a convenient single estimate that approximates to scale-specific MCIDs developed for the QLQ C15-PAL [57].
Patient empowerment
The Health Education Impact Questionnaire (heiQ) will be administered at weeks one, two and 4 to evaluate patient empowerment. A version of the heiQ developed in Australia specifically for cancer will be used. The study includes scales measuring health service navigation, constructive attitudes and approaches and skill and technique acquisition [58]. As well as offering an index of self-reported self-management efficacy, it is hypothesised from our systematic review that patient empowerment may have a direct impact on pain experience itself [23].
Carer experience
The Carer Experience Scale (CES) will be administered at weeks two and 4 to evaluate the intervention effect on unpaid carers in terms of their relationship with patients, support received and ability to carry on with their own lives [26].
Structural and process measures
The implementation strategies evaluated by this study represent a complex intervention involving multiple interacting components [18]. As such, measures of infrastructure and processes for care are needed to assess fidelity of implementation, clarify causal mechanisms and identify influential contextual factors to inform further refinement and tailoring to different settings.
Service-level process measures are: screening rates, clinician training (via spaced learning and other initiatives), adherence to the guidelines, and documentation quality.
Staff-level process measures are: the amount of time spent in training and on cancer pain screening, assessment and management by clinical staff and front desk administrative staff. Staff will be invited to complete an anonymous online survey that asks them their role at the service and three separate questions on how much time they spent on cancer pain screening, assessment and management during their last complete work day.
Patient-level process measures are: number of consultations, multi-disciplinary input, primary care support, and indices of care recommended by the guidelines. Items of particular interest will be instances of screening, assessment, patient education, regular analgesia, breakthrough analgesia and management of side-effects (preventative and treatment). The audit tool (administered by trained personnel to standardise between arms and services) will assess participants’ clinical records retrospectively. Because it is often not documented in medical records, patients’ receipt of pain education will be measured by asking the patient at each time-point they contribute to secondary data collection.
Descriptive/control variables
Patient variables will include demographics, details of cancer diagnoses, performance status (Australia - modified Karnofsky Performance Status [AKPS] [59] at palliative care services and the Eastern Oncology Cooperative Group Performance Status [ECOG] [60] at oncology services) and comorbidities that may cause pain or influence its management (e.g. renal impairment). Carer variables will include demographics and relationship to the patient. Staff variables will be limited to role.
Qualitative sub-study
A sample of patients and carers (n = 20 respectively) who participate in secondary outcome measurement will be invited to take part in interviews 4 weeks after initial screening. Interviews will use a focused question route and ask about informants’ experiences of pain assessment and management, self-management education, and perceived person-centredness and coordination of care.
On completion of the intervention arm at each service, clinicians and front desk administrative staff will be invited to participate in interviews or focus groups to explore perceived barriers and facilitators to the intervention.
Data management
A designated staff member at each site will enter baseline screening data into an Excel spreadsheet specifically developed for this study. Participants will be identified by a study number only, and data will be stored in a locked filing cabinet or on password-protected computers at the coordinating site. The investigators and sponsor will have access to the final dataset. Because the intervention arm is aimed at encouraging clinicians to provide evidence-based care rather than use a novel intervention, no data monitoring committee is required. There are no plans for independent trial audit. In the event that unexpected adverse events are associated with the conduct of the trial, incident reports will be completed by the project team and updates provided to the HREC. Participant information sheets will provide contact details for complaints. Trial results will be communicated to the research and clinical communities via journal publication and conference presentation, to which the investigators and (where appropriate) representatives from participating services will contribute as authors.
Sample size
To assess the appropriate study sample size and compute study power, we used a computer simulation allowing for 20% dropout by both services and patients. Drawing on data from 1612 consecutive patients [61], outcome data were generated to mimic pain scores we expect to see in the presenting population. We generated data from a beta distribution (scaled 0 to 1), rescaled to lie between 0 and 10, and then rounded to integer values. We then discarded data points < 2 to represent the sample of interest. We allowed service-specific means to vary between 0.05 and 0.15 to generate an appropriate intra-class correlation. We used a standard MCID of 0.5 effect size [62]. To more closely represent reality, we allowed for variation in the responses of each patient to intervention. Once a hypothetical study population had been generated, we ran a linear mixed model that included an intervention effect as well as an intervention by time effect, as well as a random service effect. We repeated this whole process several hundred times and then estimated power by computing the proportion of times that the null hypothesis would be rejected. We did this both for the main intervention effect, as well as the intervention by time effect. Two-sided hypothesis tests with type I errors of 0.05 were used. Assuming 82 patients per service at six services (N = 492), our study will have > 90% power to detect the main effect. The study will oversample by two services (i.e. a total of 8 services) and 18 patients per service (i.e. n = 100 at each service) to allow for drop-out.
Randomisation
Randomisation will be conducted at a central office by a statistician (LL) not involved in the operation of the study. A random sequence will be generated using a computerised randomisation algorithm. Allocation will be based on clusters rather than individuals, and randomisation will be concerned with the time at which each service transitions from the control arm to training and intervention. Randomisation will occur prior to data being collected at each service. Due to the nature of the intervention, concealment of allocation is not feasible. Stratification will be unnecessary because each service will serve as its own control. Patients will be automatically allocated to control or intervention arms according to the current arm of their treatment service upon their first presentation.
Randomisation will be conducted by the trial statistician (LL). Services will be enrolled by members of the project team prior to randomisation after a contract is signed by service managers. No concealment will be feasible for managers and clinicians due to the nature of the trial design and intervention. Because randomisation is concerned with the time at which each service will commence the intervention, individual patients and caregivers will be enrolled after randomisation but will be unaware of service allocation at the time of their enrolment.
Blinding
The nature of the intervention in this study renders blinding of clinicians impractical, and previous research suggests that uptake of guidelines in the control arm will be unlikely in the absence of targeted strategies of the kind to be introduced in the intervention arm [14]. Information for patients will provide only general information about the aims of the study (i.e. that it will compare different approaches to cancer pain management) rather than specifics of the design and intervention. Previous experience of cluster RCTs by the current team suggests that attempts to blind research assistants collecting data will be impractical. Instead, attention will be paid to research assistant training and standardisation of data collection as ways to limit the potential for bias. Personnel conducting analyses will be blinded to service allocation.
Statistical methods
For the main analysis, linear mixed models [63] will be used to model the outcomes of interest, while accounting for the clustering and longitudinal design. We have conducted computer simulations to confirm that this approach will work well for pain NRS scores measured on an eleven point scale. The linear mixed modeling framework is very flexible. For example, it will allow for testing whether treatment effects diminish over time. It will also allow the incorporation of additional covariates of interest, for instance patient age, gender, ethnicity and factors related to disease; and inclusion of covariates that reflect characteristics of the study services. Statistical analysis will be performed using appropriate software capable of handling data from this study design. Analyses will be repeated for all patients and patients newly referred during the intervention arm to control for the possibility that outcomes are influenced by care received prior to the intervention being implemented. For secondary patient outcomes, analyses will be repeated for patients with clinically relevant (≥2 NRS) and moderate-severe (≥5 NRS) worst and average pain.
Economic analysis
An economic evaluation will estimate the incremental costs and consequences of the intervention versus control arm. The primary outcome for the cost-effectiveness analysis will be the incremental cost per additional responder, with response defined as a clinically important improvement of 30% on a 0–10 NRS one week post-screening for those with moderate-severe (≥5 NRS) worst pain). The incremental cost per additional quality adjusted life years (QALY) will be the secondary outcome. Utility scores will be derived from the EORTC QLQ-C15-PAL and 5 additional items from the QLQ-C30 (mapped from QLQ-Utility [56]) and index values for carers from the CES. Survey responses will be linked to health service utilisation data accessed through the Centre for Health Record Linkage (CHeReL) and from Commonwealth datasets (Medicare Benefits Scheme (MBS); Pharmaceutical Benefits Scheme [PBS]) and hospital records. Variables will include: Australian Refined Diagnosis Related Groups (ARDRG) code; number of hospitalisations; length of hospital stays; number of emergency department, outpatient, psychology/psychiatry and general practitioner (GP) visits; health insurance status; medication usage. Costs of the intervention will relate to the screening systems implemented at each service, materials for training, and staff time spent on attending training and screening, assessing and managing cancer pain. As economic data may be skewed, confidence intervals will be estimated with bootstrap methods [64]. Sensitivity analysis will examine the effect of assumptions and determine which cost components drive the results.
Qualitative analysis
Interview and focus group data will be transcribed verbatim and imported into NVivo 11 software for analysis. Analysis will use an integrative qualitative method designed specifically for informing health service interventions [65]. This method uses both deductive and inductive approaches to ensure themes build on previous research whilst also remaining open to new insights from participants’ experiences. To reduce risk of bias and enrich interpretation, analysis will be conducted by two independent researchers who will then meet to agree themes. As well as coding concepts (concept codes), researchers will also code relationships between concept codes (relationship codes), participants’ positive or negative appraisals (participant perspective codes) and relevant variables relating to participant characteristics (participant characteristic codes) and service (service codes). Preliminary themes will be subject to review by the larger research team before finalizing the code structure.
The Promoting Action on Research Implementation in Health Services (PARiHS) framework will be applied to analyses of interview/focus group data and the structural and process measures outlined above to consider the influences of evidence, context and facilitation on the success of the intervention [66].
Discussion
The protocol for the Stop Cancer PAIN Trial has been designed to balance the need for rigorous evaluation with the flexibility required by complex interventions [67]. The proposed intervention is intended to take a coordinated approach to overcoming barriers to guideline implementation at system, provider and patient levels in order to ensure that care for cancer pain meets a best-practice standard across each service but can also be tailored to the needs of individual patients and families.
One of the advantages of a stepped wedge design over parallel cluster RCTs is that a diversity of services can be included without compromising comparability because each cluster serves as its own control. Inclusion of heterogeneous services in the proposed study should enable generalisability of results to a large proportion of Australian oncology and palliative care services. Our qualitative sub-study and analyses of structural and process factors will provide useful insights into the requisite conditions for the intervention to succeed and modifications needed for different service models.
The study’s methods are limited by a lack of blinding, which is necessitated by the nature of the design, intervention and outcomes. Standardised procedures for eligibility screening are designed to reduce the risk of selection bias that might arise from clinician and researcher knowledge of allocation. Logistic barriers will mean that sustainability assessment beyond the intervention arm period will be limited to audit data at the service level, rather than outcome measurement at the patient level. Sustainability of effects on patient and carer outcomes, therefore, will at best be inferred rather than directly observed. There is also a risk that practical issues relating to service availability and recruitment may make the randomisation schedule difficult to adhere to with regard to the order in which each cluster commences the control arm and transitions to the intervention [68]. The main purpose of the randomisation is to ensure that not all services commence at the same time and so limit any confounding that might occur from changes at the national level (e.g. policy) that coincide with transition from control to intervention arms. The precise order in which services commence and transition may be of lesser concern in terms of likely bias.
If implementation strategies are found to be cost-effective, they will be made available free-of-charge alongside the Australian guidelines for 'Cancer Pain Management in Adults' on the Cancer Council Australia Cancer Guidelines Wiki [16] to facilitate nationwide translation. In keeping with the continuous cycle recommended for complex interventions by the MRC, the Wiki platform will enable ongoing development and incorporation of changing evidence over time.
Other information
Registration
The trial was prospectively registered on the Australian New Zealand Clinical Trials Registry on 23rd January 2015 with the trial identification ACTRN12615000064505; World Health Organisation unique trial number U1111–1164-4649. The primary sponsor is The University of Sydney. TL is the contact for public and scientific queries: Tim Luckett; T + 61 2 9514 4861; E tim.luckett@uts.edu.au
Protocol version number and date
I001/V 5, 5th April 2017.
Roles and responsibilities
Decisions for the conduct of the trial are made by the Stop Cancer PAIN Trial Executive Committee (ML, TL, MA, JP). The trial is coordinated from IMPACCT (Improving Palliative, Aged and Chronic Care through Clinical Research and Translation) from the University of Technology Sydney. An Advisory Group meets twice yearly to identify synergies with other initiatives across Australia.
Abbreviations
- AKPS:
-
Australia - modified Karnofsky Performance Status
- ARDRG:
-
Australian Refined Diagnosis Related Groups
- CES:
-
Carer Experience Scale
- CHeReL:
-
Centre for Health Record Linkage
- CONSORT:
-
Consolidated Standards of Reporting Trials and
- ECOG:
-
Eastern Oncology Cooperative Group Performance Status
- EORTC QLQ C15-PAL:
-
European Organisation for Research and Treatment of Cancer Palliative Care
- heiQ:
-
Health Education Impact Questionnaire
- HREC:
-
Human Research Ethics Committee
- MBS:
-
Medicare Benefits Scheme
- MCID:
-
Minimal clinically important difference
- MORECare:
-
Methods for Researching End of Life Care
- MRC:
-
(UK) Medical Research Council
- NBCF:
-
(Australian) National Breast Cancer Foundation
- NRS:
-
Numerical rating scale
- PAIN:
-
Pharmacological management, anti-cancer therapy, interventional therapy for non-responsive severe pain, non-pharmacological management
- PARiHS:
-
Promoting Action on Research Implementation in Health Services
- PBS:
-
Pharmaceutical Benefits Scheme
- PDSA:
-
Plan-Do-Study-Act
- QALY:
-
Quality adjusted life years
- QOL:
-
Quality of life
- RCT:
-
Randomised controlled trial
- SD:
-
Standard deviation
- SMART:
-
Specific, measurable, achievable, relevant and time-bound
- SPIRIT:
-
Standard Protocol Items: Recommendations for Interventional Trials
References
van den Beuken-van Everdingen MH, Hochstenbach LM, Joosten EA, Tjan-Heijnen VC, Janssen DJ. Update on prevalence of pain in patients with Cancer: systematic review and meta-analysis. J Pain Symptom Manag. 2016;51(6):1070–90.e9. https://doi.org/10.1016/j.jpainsymman.2015.12.340.
Deandrea S, Montanari M, Moja L, Apolone G. Prevalence of undertreatment in cancer pain. A review of published literature. Ann Oncol. 2008;19(12):1985–91. PMC2733110
Breivik H, Cherny N, Collett B, de Conno F, Filbet M, Foubert AJ, et al. Cancer-related pain: a pan-European survey of prevalence, treatment, and patient attitudes. Ann Oncol. 2009;20(8):1420–33. https://doi.org/10.1093/annonc/mdp001.
Fortner BV, Demarco G, Irving G, Ashley J, Keppler G, Chavez J, et al. Description and predictors of direct and indirect costs of pain reported by Cancer patients. J Pain Symptom Manag. 2003;25(1):9–18. https://doi.org/10.1016/S0885-3924(02)00597-3.
Luckett T, Davidson PM, Green A, Boyle F, Stubbs J, Lovell M. Assessment and management of adult cancer pain: a systematic review and synthesis of recent qualitative studies aimed at developing insights for managing barriers and optimizing facilitators within a comprehensive framework of patient care. J Pain Symptom Manag. 2013;46(2):229–53.
Oldenmenger WH, Sillevis Smitt PAE, van Dooren S, Stoter G, van der Rijt CCD. A systematic review on barriers hindering adequate cancer pain management and interventions to reduce them: a critical appraisal. Eur J Cancer. 2009;45(8):1370–80.
Jacobsen R, Sjogren P, Moldrup C, Christrup L. Physician-related barriers to cancer pain management with opioid analgesics: a systematic review. J Opioid Manag. 2007;3(4):207–14.
Jacobsen R, Moldrup C, Christrup L, Sjogren P. Patient-related barriers to cancer pain management: a systematic exploratory review. Scand J Caring Sci. 2009;23(1):190–208.
Jacobsen R, Liubarskiene Z, Moldrup C, Christrup L, Sjogren P, Samsanaviciene J. Barriers to cancer pain management: a review of empirical research. Medicina (Kaunas). 2009;45:427–33.
Fazeny B, Muhm M, Hauser I, Wenzel C, Mares P, Berzlanovich A, et al. Barriers in cancer pain management. Wien Klin Wochenschr. 2000;112(22):978–81.
Kotronoulas G, Kearney N, Maguire R, Harrow A, Di Domenico D, Croy S, et al. What is the value of the routine use of patient-reported outcome measures toward improvement of patient outcomes, processes of care, and health service outcomes in cancer care? A systematic review of controlled trials. J Clin Oncol. 2014;32(14):1480–501. https://doi.org/10.1200/JCO.2013.53.5948.
Etkind SN, Daveson BA, Kwok W, Witt J, Bausewein C, Higginson IJ, et al. Capture, transfer, and feedback of patient-centered outcomes data in palliative care populations: does it make a difference? A Systematic Review. J Pain Symptom Manage. 2014; https://doi.org/10.1016/j.jpainsymman.2014.07.010.
Brink-Huis A, van Achterberg T, Schoonhoven L, Brink-Huis A, van Achterberg T, Schoonhoven L. Pain management: a review of organisation models with integrated processes for the management of pain in adult cancer patients. J Clin Nurs. 2008;17(15):1986–2000.
Davies P, Walker AE, Grimshaw JM. A systematic review of the use of theory in the design of guideline dissemination and implementation strategies and interpretation of the results of rigorous evaluations. Implement Sci. 2010;5:14. PMC2832624. doi:https://doi.org/10.1186/1748-5908-5-14
Luckett T, Butow PN, King MT. Improving patient outcomes through the routine use of patient-reported data in cancer clinics: future directions. Psycho-Oncology. 2009;18(11):1129–38. https://doi.org/10.1002/pon.1545.
Australian Adult Cancer Pain Management Guideline Working Party. Cancer pain management in adults. Cancer council Australia, Sydney. 2013. http://wiki.cancer.org.au/australia/Guidelines:Cancer_pain_management. Accessed 5th Oct 2014.
Lovell M, Luckett T, Boyle F, Stubbs J, Phillips J, Davidson P et al. Adaptation of international guidelines on assessment and management of cancer pain for the Australian context. Asia Pac J Clin Oncol. In Press;accepted 12th January 2015.
Craig P, Dieppe P, Macintyre S, Michie S, Nazareth I, Petticrew M et al. Developing and evaluating complex interventions: the new Medical Research Council guidance. BMJ 2008;337:a1655. Also available open-source online at www.mrc.ac.uk/complexinterventionsguidance Source: NLM. PMC2769032. doi:https://doi.org/10.1136/bmj.a1655.
Medical Research Council. Developing and evaluating complex interventions: new guidance. London: MRC; 2006.
Lovell M, Agar M, Luckett T, Davidson P, Green A, Clayton J. Australian survey of current practice and guideline use in adult cancer pain assessment and management: perspectives of palliative care physicians. J Palliat Med. 2014;10(2):e99–107. https://doi.org/10.1111/ajco.12040.
Luckett T, Davidson PM, Boyle F, Liauw W, Agar M, Green A, et al. Australian survey of current practice and guideline use in adult cancer pain assessment and management: perspectives of oncologists. Asia Pac J Clin Oncol. 2012; https://doi.org/10.1111/ajco.12040.
Phillips J, Lovell M, Luckett T, Agar M, Green A, Davidson P. Australian survey of current practice and guideline use in adult cancer pain assessment and management: the community nurse perspective. Collegian. 2014 https://doi.org/10.1016/j.colegn.2013.11.002.
Marie N, Luckett T, Davidson PM, Lovell M, Lal S. Optimal patient education for cancer pain : a systematic review and theory-based meta-analysis. J Support Care Cancer. 2013;21(12):3529–37.
Michie S, van Stralen MM, West R. The behaviour change wheel: a new method for characterising and designing behaviour change interventions. Implement Sci 2011;6:42 Source: NLM. PMC3096582. doi:https://doi.org/10.1186/1748-5908-6-42.
Lovell M, Birch M, Luckett T, Phillips J, Davidson P, Agar M et al. Screening and audit as service-level strategies to support implementation of Australian guidelines for cancer pain management in adults: a feasibility study. Pain management nursing. In Press:accepted may 13th 2018.
Al-Janabi H, Flynn TN, Coast J. Estimation of a preference-based Carer experience scale. Med Decis Mak. 2011;31(3):458–68. https://doi.org/10.1177/0272989x10381280.
Groenvold M, Petersen MA, Aaronson NK, Arraras JI, Blazeby JM, Bottomley A, et al. The development of the EORTC QLQ-C15-PAL: a shortened questionnaire for cancer patients in palliative care. Eur J Cancer. 2006;42(1):55–64.
Hussey MA, Hughes JP, Hussey MA, Hughes JP. Design and analysis of stepped wedge cluster randomized trials. Contemp Clin Trials. 2007;28(2):182–91.
Campbell MK, Elbourne DR, Altman DG. CONSORT statement: extension to cluster randomised trials. BMJ. 2004;328(7441):702–8 PMC381234. https://doi.org/10.1136/bmj.328.7441.702.
Chan A, Tetzlaff JM, Altman DG, et al. Spirit 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7. https://doi.org/10.7326/0003-4819-158-3-201302050-00583.
Murthy VH, Krumholz HM, Gross CP. Participation in cancer clinical trials: race-, sex-, and age-based disparities. JAMA. 2004;291(22):2720–6. https://doi.org/10.1001/jama.291.22.2720.
Higginson IJ, Evans CJ, Grande G, Preston N, Morgan M, McCrone P, et al. Evaluating complex interventions in end of life care: the MORECare statement on good practice generated by a synthesis of transparent expert consultations and systematic reviews. BMC Med. 2013;11:111 PMC3635872. https://doi.org/10.1186/1741-7015-11-111.
Australian Bureau of Statistics 3416.0 - Perspectives on migrants. Canberra: ABS2008.
Butow P, Bell M, Goldstein D, Sze M, Aldridge L, Abdo S, et al. Grappling with cultural differences; communication between oncologists and immigrant cancer patients with and without interpreters. Patient Educ Couns. 2011;84:398–405.
Shaw EK, Howard J, West DR, Crabtree BF, Nease DE Jr, Tutt B, et al. The role of the champion in primary care change efforts: from the State Networks of Colorado Ambulatory Practices and Partners (SNOCAP). J Am Board Fam Med. 2012;25(5):676–85 PMC3535479. https://doi.org/10.3122/jabfm.2012.05.110281.
Li LC, Grimshaw JM, Nielsen C, Judd M, Coyte PC, Graham ID. Use of communities of practice in business and health care sectors: a systematic review. Implement Sci. 2009;4(1):27. https://doi.org/10.1186/1748-5908-4-27.
Grimshaw JM, Thomas RE, MacLennan G, Fraser C, Ramsay CR, Vale L, et al. Effectiveness and efficiency of guideline dissemination and implementation strategies. Health Technol Assess. 2004;8(6):iii-iv):1–72.
Luckett T DP, Marie N, Birch MR, Stubbs J, Phillips J, Agar M, Boyle F, Lovell M. Development of patient-held resources to overcome barriers to cancer pain assessment and management. Behavioural Research in Cancer Control Conference; May 12th - 15th; Sydney2015.
Cancer Council Australia. Overcoming cancer pain: A guide for people with cancer, their families and friends. 2013. http://www.cancercouncil.com.au/wp-content/uploads/2014/01/Can487-Overcoming-Pain-NSW-Lores2.pdf. Accessed 18th February 2014.
Lovell MR, Forder P, Stockler M, Butow PN, Briganti E, Chye R, et al. A randomised controlled trial of a standardised educational intervention for patients with cancer pain. J Pain Symptom Manag. 2010;40(1):49–59.
Lovell M, Birch M-R, Luckett T, Davidson P, Green A, Marie N et al. Pain management can be achieved with commitment and support. NSW State Palliative Care Conference; October; Sydney 2014.
Kerfoot BP, Kearney MC, Connelly D, Ritchey ML. Interactive spaced education to assess and improve knowledge of clinical practice guidelines: a randomized controlled trial. Ann Surg. 2009;249(5):744–9.
Phillips JL, Heneka N, Hickman L, Lam L, Shaw T. Impact of a novel online learning module on specialist palliative care nurses' pain assessment competencies and patients' reports of pain: results from a quasi-experimental pilot study. Palliat Med. 2014;28(6):521–9. https://doi.org/10.1177/0269216314527780.
Phillips JL, Heneka N, Hickman L, Lam L, Shaw T. Can a complex online intervention improve Cancer Nurses' pain screening and assessment practices? Results from a multicenter, pre-post test pilot study. Pain Manag Nurs. 2017;18(2):75–89. https://doi.org/10.1016/j.pmn.2017.01.003.
Ivers N, Jamtvedt G, Flottorp S, Young JM, Odgaard-Jensen J, French SD, et al. Audit and feedback: effects on professional practice and healthcare outcomes. Cochrane Database Syst Rev. 2012;6:CD000259.
Dulko D, Hertz E, Julien J, Beck S, Mooney K. Implementation of cancer pain guidelines by acute care nurse practitioners using an audit and feedback strategy. J Am Acad Nurse Pract. 2010;22(1):45–55.
Lovell M, Birch M-R, Luckett T, Davidson P, Stubbs J, Phillips JL et al. Pilot of pain indicator audit tool as part of a complex intervention to improve cancer pain outcomes. World Cancer Congress; December; Melbourne 2014.
Ivers NM, Sales A, Colquhoun H, Michie S, Foy R, Francis JJ, et al. No more 'business as usual' with audit and feedback interventions: towards an agenda for a reinvigorated intervention. Implement Sci. 2014;9(14 PMC3896824) https://doi.org/10.1186/1748-5908-9-14.
New South Wales Health. The Clinican's Toolkit For Improving Patient Care. Australia: New South Wales Health; 2001.
Hjermstad MJ, Fayers PM, Haugen DF, Caraceni A, Hanks GW, Loge JH, et al. Studies comparing numerical rating scales, verbal rating scales, and visual analogue scales for assessment of pain intensity in adults: a systematic literature review. J Pain Symptom Manag. 2011;41(6):1073–93.
Cleeland CS, Ryan KM. Pain assessment: global use of the brief pain inventory. Ann Acad Med Singap. 1994;23(2):129–38.
Serlin RC, Mendoza TR, Nakamura Y, Edwards KR, Cleeland CS. When is cancer pain mild, moderate or severe? Grading pain severity by its interference with function. Pain. 1995;61(2):277–84.
Basch E, Trentacosti AM, Burke LB, Kwitkowski V, Kane RC, Autio KA, et al. Pain palliation measurement in cancer clinical trials: the US Food and Drug Administration perspective. Cancer. 2014;120(5):761–7. https://doi.org/10.1002/cncr.28470.
Norman GR, Sloan JA, Wyrwich KW. The truly remarkable universality of half a standard deviation: confirmation through another look. Expert Review of Pharmacoeconomics & Outcomes Research. 2004;4(5):581–5.
Aaronson NK, Ahmedzai S, Bergman B, Bullinger M, Cull A, Duez NJ, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85:365–76.
Rowen D, Brazier J, Young T, Gaugris S, Craig BM, King MT, et al. Deriving a preference-based measure for cancer using the EORTC QLQ-C30. Value Health. 2011;14(5):721–31. https://doi.org/10.1016/j.jval.2011.01.004.
Bedard G, Zeng L, Zhang L, Lauzon N, Holden L, Tsao M, et al. Minimal important differences in the EORTC QLQ-C15-PAL to determine meaningful change in palliative advanced cancer patients. Asia Pac J Clin Oncol. 2013; https://doi.org/10.1111/ajco.12069.
Maunsell E, Lauzier S, Brunet J, Pelletier S, Osborne RH, Campbell HS. Health-related empowerment in cancer: validity of scales from the health education impact questionnaire. Cancer. 2014;120(20):3228–36. https://doi.org/10.1002/cncr.28847.
Abernethy A, Shelby-James T, Fazekas B, Woods D, Currow D. The Australia-modified Karnofsky performance status (AKPS) scale: a revised scale for contemporary palliative care clinical practice. BMC Palliative Care. 2005;4(7) http://www.biomedcentral.com/content/pdf/1472-684X-4-7.pdf.
Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, et al. Toxicity and response criteria of the eastern cooperative oncology group. Am J Clin Oncol. 1982;5:649–55.
Yennurajalingam S, Kang JH, Hui D, Kang DH, Kim SH, Bruera E. Clinical response to an outpatient palliative care consultation in patients with advanced cancer and cancer pain. J Pain Symptom Manag. 2012;44(3):340–50. https://doi.org/10.1016/j.jpainsymman.2011.09.014.
Norman GR, Sloan JA, Wyrwich KW. Interpretation of changes in health-related quality of life: the remarkable universality of half a standard deviation. Med Care. 2003;41:582–92.
Fitzmaurice G, Laird N, Ware JE. Applied longitudinal analysis. New Jersey, US: John Wiley & Sons; 2004.
Briggs AH, Wonderling DE, Mooney CZ. Pulling cost-effectiveness analysis up by its bootstraps: a non-parametric approach to confidence interval estimation. Health Econ. 1997;6(4):327–40.
Bradley EH, Curry LA, Devers KJ. Qualitative data analysis for health services research: developing taxonomy, themes, and theory. Health Serv Res. 2007;42(4):1758–72.
Stetler CB, Damschroder LJ, Helfrich CD, Hagedorn HJ. A guide for applying a revised version of the PARIHS framework for implementation. Implement Sci : IS. 2011;6:99- doi:https://doi.org/10.1186/1748-5908-6-99.
Eccles MP, Armstrong D, Baker R, Cleary K, Davies H, Davies S, et al. An implementation research agenda. Implement Sci. 2009;4(18 PMC2671479) https://doi.org/10.1186/1748-5908-4-18.
Hemming K, Haines TP, Chilton PJ, Girling AJ, Lilford RJ. The stepped wedge cluster randomised trial: rationale, design, analysis, and reporting. BMJ. 2015;350:h391. https://doi.org/10.1136/bmj.h391.
Acknowledgements
The authors wish to acknowledge Prof Ian Olver and Jutta von Dincklage for hosting the pain guidelines on the Cancer Council of Australia Cancer Guideline Wiki. We also acknowledge contributions to developing the implementation strategies to be evaluated in this trial by Natalie Marie, Mary-Rose Birch, Anna Green and Dr. Ben Forster. Dr. Ben Forster and author Prof Fran Boyle were supported by the Friends of the Mater Foundation. We acknowledge members of the trial’s Executive Committee responsible for overseeing the project, including: A/Prof Melanie Lovell (Chair), Prof Meera Agar, Dr. Tim Luckett and Prof Jane Phillips. We acknowledge project team members at the coordinating centre (University of Technology Sydney) responsible for recruiting participants and collecting and managing data, including: Annmarie Hosie, Tim Luckett, Alison Read (project managers), Molly Cao, Sally Fielding, Layla Hall, Renee Xu (research assistants), Seong Cheah, Kaniz Fatema (data managers). Finally, we acknowledge members of the Stop Cancer PAIN Advisory Group for their ongoing support and advice to the project, including: Prof Sanchia Aranda, Prof Phyllis Butow, Dr. Ben Forster, A/Prof Michael Izard, Ms. Niamh O’Neill, Dr. Nathan Taylor, Dr. Ian Thong, Ms. Noelene Trotter, Ms. Jutta von Dincklage and Prof Patsy Yates.
Funding
The trial is funded by the Australian National Breast Cancer Foundation. Contact details are as follows: Level 9, 10 Barrack Street, Sydney, NSW 2000, Australia; T: + 61 2 8098 4800 F: + 61 2 8098 4801 E: info@nbcf.org.au The sponsor has had no role in the study design beyond requesting an over-sampling of patients with breast cancer, and has no role in: the collection, management, analysis, and interpretation of data; writing of the report; and the decision to submit the report for publication.
Availability of data and materials
The datasets generated and/or analysed during the current study will be made available in an online repository via RedBox.
Author information
Authors and Affiliations
Contributions
All authors (TL, JP, MA, LL, PMD, NM, FB, TS, DCC, AR, AH, ML) contributed to the development and reporting of this protocol. LL conducted the sample size estimate and statistical analysis plan. All authors (TL, JP, MA, LL, PMD, NM, FB, TS, DCC, AR, AH, ML) have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval has been granted by the South Western Sydney Local Health District Human Research Ethics Committee (HREC) – ethics approval number HREC/14/LPOOL/479. Approval has been granted by the HREC for an opt-out procedure to contact patients at Week one as well as a procedure to obtain informed verbal (rather than written) consent to participate. Verbal consent was considered to place less burden on patients who, due to their illness, might be less able to return written consent forms by mail. Written informed consent will be required on a standardised Medicare form to access PBS and MBS data.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Luckett, T., Phillips, J., Agar, M. et al. Protocol for a phase III pragmatic stepped wedge cluster randomised controlled trial comparing the effectiveness and cost-effectiveness of screening and guidelines with, versus without, implementation strategies for improving pain in adults with cancer attending outpatient oncology and palliative care services: the Stop Cancer PAIN trial. BMC Health Serv Res 18, 558 (2018). https://doi.org/10.1186/s12913-018-3318-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12913-018-3318-0