
Abstract
Background
With the growing amount of clinical research, regulations and research ethics are becoming more stringent. This trend introduces a need for quality assurance measures for ensuring adherence to research ethics and human research protection beyond Institutional Review Board approval. Audits, one of the most effective tools for assessing quality assurance, are measures used to evaluate Good Clinical Practice (GCP) and protocol compliance in clinical research. However, they are laborious, time consuming, and require expertise. Therefore, we developed a simple auditing process (a screening audit) and evaluated its feasibility and effectiveness.
Methods
The screening audit was developed using a routine audit checklist based on the Severance Hospital’s Human Research Protection Program policies and procedures. The measure includes 20 questions, and results are summarized in five categories of audit findings. We analyzed 462 studies that were reviewed by the Severance Hospital Human Research Protection Center between 2013 and 2017. We retrospectively analyzed research characteristics, reply rate, audit findings, associated factors and post-screening audit compliance, etc.
Results
Investigator reply rates gradually increased, except for the first year (73% → 26% → 53% → 49% → 55%). The studies were graded as “critical,” “major,” “minor,” and “not a finding” (11.9, 39.0, 42.9, and 6.3%, respectively), based on findings and number of deficiencies. The auditors’ decisions showed fair agreement with weighted kappa values of 0.316, 0.339, and 0.373. Low-risk level studies, single center studies, and non-phase clinical research showed more prevalent frequencies of being “major” or “critical” (p = 0.002, < 0.0001, < 0.0001, respectively). Inappropriateness of documents, failure to obtain informed consent, inappropriateness of informed consent process, and failure to protect participants’ personal information were associated with higher audit grade (p < 0.0001, p = 0.0001, p < 0.0001, p = 0.003). We were able to observe critical GCP violations in the routine internal audit results of post-screening audit compliance checks in “non-responding” and “critical” studies upon applying the screening audit.
Conclusions
Our screening audit is a simple and effective way to assess overall GCP compliance by institutions and to ensure medical ethics. The tool also provides useful selection criteria for conducting routine audits.
Similar content being viewed by others


Background
Good Clinical Practice (GCP) is an international scientific and ethical quality standard for designing, conducting, recording, and reporting clinical research using human participants. It originated from the Declaration of Helsinki [1]. Clinical research using human participants should ensure GCP compliance throughout the research period. Investigators are responsible for ensuring that the research activities achieve initial or continuing approval from the relevant Institutional Review Board (IRB). In addition, an independent method for confirming whether research complies with the relevant laws and regulations is an audit. An audit is a systematic monitoring method used to determine whether clinical-research-related activities and documents are adequately obtained according to the approved protocol, GCP, and the applicable regulatory requirements.
In recent years, the scope and amount of clinical research has expanded, and regulations are becoming more stringent [2, 3]. Increasingly, human research protection programs (HRPPs) are being introduced by research organizations to promote ethical values in the research community and to ensure an environment of participant protection. As a component of HRPP, an audit confirms all research documents and procedures in regards to whether they follow regulations and protocol. Auditing procedures, however, are not easy for most organizations, and encompass preparing an audit’s standard of procedure (SOP), protocol selection, recording the audit trail, source document verification, and investigator interviews, which require approximately three to 4 days and expert staff. Furthermore, an internal audit cannot cover all active protocols because of time limits. Accordingly, audits are time consuming and can be a laborious burden for both organizations initially adopting an HRPP and organizations already operating one. Seeing an unmet need for measures with which to ensure quality compliance by research organizations, we developed a “screening audit” that confirms whether trial documents have been properly set aside and whether all active protocols are being adhered to: it does this without interviews or source document verification. The aim of this study was to evaluate the effectiveness of the screening audit in confirming GCP compliance status among investigators and research institutions.
Methods
Development of the screening audit
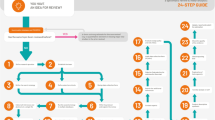
This study was conducted as a preliminary exploratory study through which to introduce the screening audit and to provide information on its effectiveness based on data from a single institution. The scope of an audit can vary according to the institution or sponsor’s SOP. While routine audit is a scrutinizing way to confirm GCP compliance status, it is also laborious and time consuming. A detailed routine audit consists of 10 steps, and the screening audit is applicable to the step of trial master file (TMF) review (Fig. 1). At the opening meeting, the auditor explains the target, scope, and purpose of the audit, and may ask the investigator to give an overall explanation about the protocol or GCP in order to evaluate the investigator’s understanding thereof. The primary areas of concern are the availability of essential documents and the appropriateness and completeness of documentation.

Scheme of detailed process of a routine audit and the scope of a ‘screening audit’
We developed the screening audit based on the internal audit procedure of Severance Hospital. The screening audit is performed only with available documents, without interviews, response feedback, or data accuracy evaluations, as shown in Table 1. The checklist was composed of 20 questions grouped into five audit-finding categories (Table 2). Instead of an IRB decision on the audit findings and response, auditors themselves make a decision on each protocol based on the findings from the screening audit. Both routine audit and screening audit should follow the ICH-GCP [1], U.S. Food and Drug Administration (FDA) guidance [4], and Korean GCP.
Screening audit was performed annually for all ongoing clinical research, including clinical trials with investigational drugs/devices, biospecimen research, social behavioral research, etc. The relevant IRB should determine a protocol’s risk level (out of four possible grades) based on a risk and benefit analysis. Risk level 1 is minimal risk, which is encountered in daily life or at a routine doctor’s visit. Risk level 2 means minor increase over minimal risk, and risk level 3 goes to possibility of significant sequelae, must be transient. Risk level 4 is defined when the research subjects could experience death or irreversible, debilitating damage, or major unknowns with respect to the intervention.
Audit findings were presented as a numeric count. Auditors classified findings into four different grades, including ‘Not a finding’, ‘Minor’, ‘Major’, and ‘Critical’. According to Korea FDA guideline and Severance Hospital Human Research Protection Program Policy and Procedure (HRPP P&P), ‘not a finding’ means a protocol in which there are no findings, while ‘Minor’ decision was made when complementary measures are necessary to ensure participant safety and reliability of the research ethics. ‘Major’ is defined as findings that are likely to put a participant at risk or to hinder the quality of the research, while ‘Critical’ is defined as findings significantly affecting the reliability of the research results, especially when major findings are continuously or repeatedly identified in the routine audit. In case of screening audit, protocols with more than three missing documents or inappropriate consent processes or failure to obtain informed consent forms were defined as ‘Critical’; more than two missing documents was defined as ‘Major’; and one missing documents was defined as ‘Minor’. Finally, protocols in which no problems are detected are defined as ‘Not a finding’.
Materials
We retrospectively reviewed the screening audit records of 462 protocols regularly assessed by the Human Research Protection Center at Severance Hospital, Yonsei University College of Medicine, Seoul, Korea, between January 2013 and January 2017. Protocol inclusion criteria for the screening audit were all active protocols approved by Severance Hospital’s IRB, with greater focus on investigator-initiated trials, prospective studies, and the protocols that have not undergone routine audits. An IRB was established in Severance Hospital in 1997, and all active protocols approved by the IRB since its establishment were included. The screening audit was conducted by five qualified auditors with more than 2 years of experience in the Human Research Protection Center quality assurance (QA) team. In order to evaluate the validity of the audit results, the auditors cross-checked each other’s audit findings with blind study information and made an assessment of their own. An assessment of the results’ validity was conducted with 265 protocols between January 2013 and January 2014.
Statistical method
All analyses were conducted using SAS version 9.4 (SAS Institute Inc., Cary, NC) or SPSS 23.0 for Windows (SPSS Inc., Chicago, IL), with a p-value < 0.05 being considered statistically significant. Descriptive data are expressed as a mean ± standard deviation and categorical variables as a frequency and percentage. Differences between screening audit results according to year, risk level, and department were analyzed with Chi-square test and Fisher’s exact test. Validation between results evaluated by three individual auditors were sought using the weighted kappa. Finally, a comparison of the audit findings among different grading groups or finding categories were conducted using the Chi-square test. To confirm the ordinal variation, Mantel-Haenszel Chi-square test was conducted.
Results
Baseline characteristics of protocols
Table 3 shows the basic characteristics of the protocols that underwent screening audit. Most protocols involved non-phase clinical research (n = 361, 78.1%), although we also covered phase 1 to 4 clinical trials. The majority exhibited minimal risk (n = 263, 56.9%), 152 (32.9%) were level 2, 31 (6.7%) were level 3, and one (0.2%) was level 4. The study determined to be risk level 4 was a prospective clinical study involving a surgical procedure for an aortic stenosis patient proposed by cardiology investigators, and the IRB analyzed the anticipated mortality risk in comparison with other standard surgical procedures. The prevalent characteristics of the studies were “conducted at a single center” (n = 281, 60.8%) and academic purpose (n = 455, 98.1%). Principle investigators who had experienced screening audits mainly came from the Department of Internal Medicine (n = 179, 38.7%).
Investigator compliance according to reply rate
The annual reply rates in the screening audits are shown in Fig. 2. Overall, 73% of the investigators submitted TMF for the first screening audit in 2013. Successively, reply rates of 26, 53, 49, and 55% were recorded in the following years. The reason for the large number of candidate protocols in January 2013 was that it was the first screening audit experience, and therefore, included all active protocols approved by the IRB since its establishment. In comparison with the average number of initial reviews in the IRB, approximately 10% are required for screening audit annually.

Annual reply rate for screening audits. The screening audit involved a request to the researchers for voluntary submission, and unresponsive researchers could be considered as candidates for subsequent internal audit
Validation of auditor decisions
Since a screening audit primarily focuses on mandatory trial documents, not the content, it is possible to make a relatively objective evaluation. Based on the individual audit checklist, three different auditors gave a grade for the reliability test. The auditors sorted the audit results into four grades (“not a finding,” “minor,” “major”, and “critical”) using the screening audit checklist. The three independent auditors made a decision using the system “Not a finding”=1, “minor” = 2, “major” = 3, and “critical” = 4. The means and standard deviations of the three auditors were 2.46 ± 0.84, 2.35 ± 0.69, and 2.55 ± 0.70, respectively. Weighted kappa values derived from three different pairs of auditors were 0.316 [CI:0.285,0.348], 0.339 [CI:0.305,0.373], and 0.373 [CI:0.342,0.404]), showing fair agreement (weighted kappa of 0.21–0.40) [5].
Audit outcomes and associated factors
In our study, 55 protocols (11.9%) were “critical,” 180 (39.0%) were “major,” 198 (42.9%) were “minor,” and 29 (6.3%) were “not a finding.” Differences in grade according to year were not statistically significant (p = 0.813). Frequencies of protocols determined as “not a finding” and “minor” gradually increased from risk levels 1 to 3 (4.9% → 7.9% → 12.9% and 39.9% → 44.1% → 61.3%, respectively), while frequencies of “major” and “critical” protocols were significantly higher (at 41.1% and 14.1%, respectively) for risk level 1 than risk levels 2 (38.2%, 9.9%) and 3 (22.6%, 3.2%) (p = 0.002). International multicenter studies had higher frequencies of “not a finding” than domestic multicenter or single center studies. In contrast, single center studies had higher proportions of “critical” and “major” than domestic multicenter and international multicenter studies (p < 0.0001). According to the responsible entity of the study, grade was not statistically different (p = 0.625). Also, non-phase clinical research had more “critical” and “major” protocols than phase-specific research that included phase 1, 1/2, 2, 2/3, 3, and 4, as well as Post Market Surveillance and medical-device trials (14.13%, 42.11% > 3.96%, 27.72%) (p < 0.0001). Protocols performed by basic science departments had significantly higher frequencies of a “critical” finding as 21.4% than those by clinical departments (Internal Medicine (7.3%), supportive departments (9.9%), Surgical departments (15.6%), p = 0.001) (Table 4).
Audit findings
We categorized the audit findings into five types: 1) Failure to maintain essential documents, 2) inappropriateness of documents, 3) failure to obtain informed consent, 4) inappropriateness of informed consent form, and 5) failure to protect participants’ personal information. The findings were collected as numeric counts. The number of findings was related to different types of questions in the screening audit checklist. The severity or proportion of failure to total participants was not reflected. Table 5 shows the association between grades and finding categories. Higher frequencies of “inappropriateness of documents” were prevalent in “not a finding” and “minor” findings (p < 0.0001). Also, higher frequencies of “failure to obtain informed consent” and “inappropriateness of informed consent form” were associated with audit result of higher grade (p = 0.0001 and p < 0.0001, respectively). When auditors found that informed consent was either not obtained or had undergone a false consent process, they tended to regard it as serious non-compliance. In contrast, “failure to maintain essential documents” was not significantly associated with grade (p = 0.091).
Routine audit as a compliance monitoring method of screening audit
If serious non-compliance was detected in any protocols through a screening audit, they could have been subjected to routine audit. During our study period, 29 protocols received subsequent routine audits due to issues that arose from the screening audit. Specifically, 12 “non-responding” studies and 17 studies determined as “critical” in the screening audit were involved. Two studies were determined as “major,” although the auditors considered it is necessary to perform subsequent monitoring via routine audit. In a compliance monitoring process subsequent to the screening audit, 17.2% of protocols were determined as “critical,” the same as that in the screening audit; 44.8% were determined as “major”; and 37.9% were “minor” in the final IRB decision. Differences between the “critical, major” group and the “non-responding” group were not significant (Table 6).
Discussion
Laws and regulations related to clinical research have become more stringent in response to the dramatic increase in the number and diversity of clinical research during recent decades [6]. These changes have made QA more important [7]. QA activities intending to improve research quality may involve inspection, sponsor monitoring, and audits of research institutions [8]. Audits are routinely conducted to confirm whether clinical research has been performed in accordance with regulations and the protocol. Audits are a relatively independent and vigorous QA activity; however, they are laborious and time-consuming. The reason for this is that the auditor must be well acquainted with relevant regulations and the study’s procedure. Furthermore, a sufficient training period is needed for the same reason [9]. Thus, the authors developed the novel screening audit as a simple and easy method for HRPPs. The screening audit is executed to grasp the overall management status of clinical research conducted after IRB approval is granted and to confirm the appropriate management of essential documents. It can be conducted using a yes/no checklist, such that auditors without sufficient experience can also be involved, as shown in Table 2.
One of the most important advantages of the screening audit is that it is very easy to adapt to a real-world setting. The checklist consists of 20 questions extracted from a routine audit checklist, with one being that an essential documents list should be maintained. It has no source document or medical record verification or investigator interviews, and it only takes about 30 min to 2 h to complete, which is much shorter than a routine audit. Secondly, a screening audit is an opportunity to inform all investigators about essential trial documents. Because every investigator who is conducting ongoing clinical research is a candidate, it can cover almost every investigator in the institution. With the screening audit, almost every investigator can be informed about clinical research QA activity and basics of study document composition. Furthermore, a routine audit can only detect problems with a randomly chosen protocol, while a screening audit can assess overall research compliance in an institution. In the baseline characteristics of screening audit protocols, phase 1 to 4 clinical trials, post-market surveillance, research with medical devices, and non-phase clinical research are all covered. It was possible for us to observe investigators’ perceptions of QA activity within HRPPs, which have gradually improved, as seen in the annual reply rate. Also, a screening audit can cover the blind spots in a routine audit, such as minimal risk studies or studies conducted by basic science departments.
In recent decades, audits have become more effective in several ways. Califf et al. [10] first provided a cost-effective auditing system in 1997; thereafter, risk analysis has tended to be adopted in research monitoring systems [11]. Most recently, FDA announced guidelines regarding a risk-based approach to monitoring [12]. Risk-based monitoring uses several factors to select protocols, such as the complexity of the study design, the types of endpoint, the clinical complexity of the study population, the relative experience of the investigator, etc. As a result of the screening audit, studies with risk levels 1–2 or conducted by basic science departments or supportive departments were observed as having more “major” or “critical” findings than risk level 3–4 studies or those conducted by departments of internal medicine or surgery. This may indicate the needs for investigator training in basic science departments and supportive departments, as well as low-risk studies.
Previous research assessed U.S. FDA inspection results from 1977 to 2009 and provided the distribution of audit findings for FDA-approved clinical trials [13]. In this article, “failure to follow investigational plan” was frequently observed, at 33.8%. The remaining results were as follows: 28.1% for “inappropriateness of informed consent procedure,” 27.0% for “inappropriateness of documentation,” 15.2% for “insufficient management of investigational product,” and 5.9% for “unreported adverse events.” In our study, “failure to maintain essential documents” was the most frequently observed, while “failure to obtain informed consent forms” was relatively rare. We assumed that the difference had resulted from the screening audit’s focus on documents. However, findings of any misconduct in the informed consent procedure were key factors in determining the grade as “major” or “critical.” Since an appropriate consent procedure is more crucial than minor procedural errors, such as the failure to maintain current versions of study documents in terms of human participant protection [12, 14].
Screening audit can be used as an effective selection tool for routine audit. In this article, we defined investigator non-compliance as “non-responding” and “critical.” Those studies showed high rates of “critical” and “major” grades in the final IRB decision after the internal audit was conducted. “Critical” (17.2%) and “major” (44.8%) grades were relatively frequent for non-compliant studies that needed further routine audit. Taking these findings together, we suggest that “critical” or “non-responding” studies should be regarded as primary candidates for routine audit to ensure a scrupulous review. Furthermore, screening audits are limited when it comes to checking the appropriateness of documentation, which enables an objective evaluation. Auditing is a complex procedure and it changes according to study characteristics [10]. We analyzed the results of audit grading reliability on the same studies by three different auditors, and found a fair degree of agreement.
This study has several limitations. First, it was a retrospective study that was based on records from an audit database. Nevertheless, we were able to develop the concept of screening audits by performing a well-organized, statistical analysis with a grading index. Second, the data were collected from a single tertiary hospital that was running an HRPP. This might limit the generalizability of our data to the general research environment. However, our institution has approximately 1500 ongoing studies, which is highest in Korea, so we believe that the bias would be minimal.
Conclusion
Screening audits are an effective method for overseeing and controlling an institution’s overall GCP compliance, and can be useful selection criteria for a routine audit. The usefulness of screening audits has not yet been established because of insufficient evidence. However, based on our study, this new screening audit seems to be an effective monitoring method in the challenging environment of clinical research ethics and its oversight, where the increasing variability and magnitude of clinical research make things difficult. Further, multi-center, multi-national studies are needed to validate our conclusion.
Abbreviations
- FDA:
-
Food and Drug Administration
- GCP:
-
Good Clinical Practice
- HPC:
-
Human research Protection Center
- HRPP:
-
Human Research Protection Program
- ICF:
-
Informed consent form
- IRB:
-
Institutional Review Board
- PMS:
-
Post market surveillance
- QA:
-
Quality assurance
- TMF:
-
Trial master files
References
Guideline IH. Integrated addendum to ICH E6 (R1): guideline for good clinical practice E6 (R2). Current Step. 2015;2:1–60.
Medicine Io. Contemporary issues for protecting patients in cancer research: workshop summary. Washington, DC: The National Academies Press; 2014.
Speers MA. Making human research safe: why we cannot afford to fail. Sci Eng Ethics. 2005;11(1):53–9.
Lisook AB. FDA audits of clinical studies: policy and procedure. J Clin Pharmacol. 1990;30(4):296–302.
Fleiss JL. Statistical methods for rates and proportions. New York: Wiley; 1981.
Park S, Noh YH, Rha SY, Kim WH, Cheon JH. Institutional board review for clinical investigations on inflammatory bowel diseases: a single-center study. Intest res. 2015;13(3):274–81.
Glickman SW, McHutchison JG, Peterson ED, Cairns CB, Harrington RA, Califf RM, et al. Ethical and scientific implications of the globalization of clinical research. NEJM. 2009;360(8):816–23.
Bhatt A. Quality of clinical trials: a moving target. Perspect Clin Res. 2011;2(4):124–8.
Hattemer-Apostel R. GCP auditors: hard to find—hard to develop—hard to keep: I. criteria and methods for candidate selection. Qual Assur J. 2000;4(1):3–8.
Califf RM, Karnash SL, Woodlief LH. Developing systems for cost-effective auditing of clinical trials. Control Clin Trials. 1997;18(6):651–60.
Brosteanu O, Houben P, Ihrig K, Ohmann C, Paulus U, Pfistner B, et al. Risk analysis and risk adapted on-site monitoring in noncommercial clinical trials. Clin Trials. 2009;6(6):585–96.
Food, Administration D. Guidance for industry: oversight of clinical investigations—a risk-based approach to monitoring. Silver Spring: FDA; 2013.
Morgan-Linnell SK, Stewart DJ, Kurzrock R. U.S. Food and Drug Administration inspections of clinical investigators: overview of results from 1977 to 2009. Clin Cancer R. 2014;20(13):3364–70.
Baigent C, Harrell FE, Buyse M, Emberson JR, Altman DG. Ensuring trial validity by data quality assurance and diversification of monitoring methods. Clin Trials. 2008;5(1):49–55.
Funding
This study was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (1520190).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors read and approved the final manuscript to be published. SYR contributed to research question and design and contributed to data collection. Study concept and design: SinyoungP, YHN, CRA, WSY, BKK, SMK, JSK, SYR. Analysis and interpretation of data: SinyoungP, CMN, SejungP, SYR.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval and participant consent was not necessary as this study involved the use of an encrypted database and does not contain personal information or identifiers according to the Bioethics and Safety Act, Korea.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Park, S., Nam, C.M., Park, S. et al. ‘Screening audit’ as a quality assurance tool in good clinical practice compliant research environments. BMC Med Ethics 19, 30 (2018). https://doi.org/10.1186/s12910-018-0269-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12910-018-0269-2