
Abstract
Background
Laccases are multicopper oxidases, which are assigned into auxiliary activity family 1 (AA1) in the CAZy database. These enzymes, catalyzing the oxidation of phenolic and nonphenolic substrates coupled to reduction of O2 to H2O, are increasingly attractive as eco-friendly oxidation biocatalysts. Basidiomycota laccases are well characterized due to their potential in de-lignification of lignocellulose. By contrast, insight into the biochemical diversity of Ascomycota counterparts from saprophytes and plant pathogens is scarce.
Results
Here, we report the properties of the laccase from the major wheat pathogen Zymoseptoria tritici (ZtrLac1A), distinguished from common plant fungal pathogens by an apoplastic infection strategy. We demonstrate that ZtrLac1A is appended to a functional starch-binding module and displays an activity signature disfavoring relatively apolar phenolic redox mediators as compared to the related biochemically characterized laccases. By contrast, the redox potential of ZtrLac1A (370 mV vs. SHE) is similar to ascomycetes counterparts. The atypical specificity is consistent with distinctive sequence substitutions and insertions in loops flanking the T1 site and the enzyme C-terminus compared to characterized laccases.
Conclusions
ZtrLac1A is the first reported modular laccase appended to a functional starch-specific carbohydrate binding module of family 20 (CBM20). The distinct specificity profile of ZtrLac1A correlates to structural differences in the active site region compared to previously described ascomycetes homologues. These differences are also highlighted by the clustering of the sequence of ZtrLac1A in a distinct clade populated predominantly by plant pathogens in the phylogenetic tree of AA1 laccases. The possible role of these laccases in vivo merits further investigations. These findings expand our toolbox of laccases for green oxidation and highlight the binding functionality of CBM-appended laccases as versatile immobilization tags.
Similar content being viewed by others


Background
Laccases (EC 1.10.3.2) are multicopper oxidases (MCOs), which catalyze monoelectric oxidation of a variety of substrates using molecular oxygen that is reduced to water [1,2,3]. A mononuclear copper binding site designated as a type-1 Cu (blue Cu) and a trinuclear copper site, involving one type-2 and two type-3 Cu (coupled binuclear Cu) binding sites are typically present in laccases. Substrates are oxidized in vicinity of the high redox potential mononuclear site (Cu1 or T1), which is responsible for the blue color and distinctive absorbance of these enzymes at about 600 nm [1, 4, 5]. Thus, the type-1 Cu possesses a higher redox potential as compared to its counterparts in the type 2 and type 3 sites [6]. The electrons from the oxidation of substrates at the T1 site are transferred through the protein via the Cys-His pathway to the trinuclear cupper cluster at the T2/T3 sites, where the electrons are transferred to oxygen (O2) [5].
Laccases are considered as eco-friendly catalysts due to their ability to oxidize a broad range of organic and synthetic substrates using only oxygen as the electron acceptor with no toxic byproducts. Some potential substrates are large in size precluding their accommodation into the active sites of laccases. Small redox mediators, e.g. 2,2-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS), 2,6 dimethoxyphenol, DMP and syringaldazine (Fig. 1), generally act as electron shuttles that mediate the oxidation of substrates too large for the direct oxidation.

Chemical structures of three synthetic redox mediators, a 2,2-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt, ABTS (b) 2,6 dimethoxyphenol, DMP (c) syringaldazine
Although, laccases are produced by diverse taxonomic groups including bacteria, insects, lichens, plants and fungi, the latter category mainly dominates in biotechnological applications. The wide utilization of fungal laccases in biotechnological applications is likely attributed to the stability, industrial efficient heterologous expression systems and high redox potential of Basidiomycota enzymes [3, 7]. Particularly, the abundance of laccases in almost all wood-rotting fungi has attracted interest in the context of de-lignification of lignocellulose, but laccases are also implicated in lignification, oxidative stress management in plants and are frequently active on an array of phenolic compounds (e.g. phenols, polyphenols, benzenothiols and anilines) [8].
Fungal laccases are produced by several groups ranging from yeasts, white and brown rot fungi (both from the Basidiomycota phylum), or Ascomycota as well as mycorrhizal species. Laccases are classified into auxiliary activity family 1 (AA1) in the Carbohydrate-Active Enzyme (CAZy) database [9]. Laccases from white rot basidiomycetes are distinguished by their high redox potentials (E0 ≈ 0.8 V for the Cu1 site) [10], as compared to ascomycete and/or bacterial laccases, which display redox potential E0 = 0.4–0.7 V [11]. Notably, the genomes of phytopathogenic ascomycete fungi like e.g. Botrytis cinera [12], Magnaporthe grisea [13] and Fusarium oxysporum [14] each encode several putative laccases of AA1 subfamily 3 (AA1_3), which harbors most ascomycete laccases according to the CAZy classification, whereas basidiomycetes counterpart are assigned into AA1 subfamily 1 (AA_1). Functional insight into AA1_3 laccases from phytopathogens, which may contribute to the understanding of the properties and possible roles of these enzymes during pathogenesis, lags behind.
Zymoseptoria tritici (syn. Mycosphaerella graminicola, Septoria tritici) is responsible for the wheat disease Septoria tritici blotch (STB) [15, 16]. This plant disease has been recognized as the most devastating for wheat production in Europe [17, 18], which is also highlighted by the ranking of the causative fungus amongst the top ten fungal pathogens [19]. Z. tritici is distinguished by an infection within the apoplastic space (outside the plasma membrane but within the plant cell wall) that differentiates it from other globally recognized fungal phytopathogens e.g. Fusarium [20] and Magnaporthe spp. [21]. The genome of M. graminicola IPO323, reveals a significant reduction in the number of carbohydrate active enzymes (CAZymes) that target plant cell walls, especially those possessing a carbohydrate binding module (CBM) [22], consistent with this apoplastic infection strategy. Remarkably, starch-binding modules of CBM family 20 (CBM20) are encoded by four genes from Z. tritici: one encoding a hypothetical protein, two encoding putative α-glucosidases and a fourth encoding a laccase.
In this study, we produced and characterized the first modular laccase (hereafter referred to as ZtrLac1A) comprising a catalytic module of AA1_3 and a starch-binding module of CBM20. Curiously, this enzyme has a preference to the negatively charged redox mediator 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) as compared to more apolar mediators. This activity profile is the converse of fungal laccases of AA1_3 characterized to date.
To provide a rationale for this different specificity, we carried out bioinformatic analyses, which revealed marked differences between ZtrLac1A and characterized counterparts in the vicinity of the T1 site, highlighting this region as an important specificity determinant. Moreover, variations in sequence were also observed at the C-terminus, which has been shown to be an important activity signature in laccases. The modularity and atypical specificity profile of ZtrLac1A offer an expansion of our enzymatic tool box of green oxidative enzymes and promote our understanding of the basic features that govern the specificity of AA1 laccases.
Methods
Materials and media
ABTS, syringaldazine and 2,6 DMP (2,6-dimethoxyphenol) and other chemicals were from Sigma-Aldrich (St. Louis, MO, USA). Restriction enzymes and molecular biology reagents were from New England Biolabs (Ipswich, MA, United States). All chemicals were of analytical grade. The Myrothecium verrucaria bilirubine oxidase (MvBOD, 3.61 mg/mL) was from Novozymes (Bagsværd, Denmark). The osmium polymer ([Os (4,4′-dichloro-2,2′-bipyridine)2(poly-vinylimidazole)10Cl]·Cl, E°′ = 0.350 V vs. Ag|AgClsat) (Additional file 1: Figure S1) was kindly provided by Prof. Dónal Leech and Dr. Peter Ó Conghaile from Biomolecular Electronics Research Laboratory, National University of Ireland (Galway, Ireland) and prepared as previously reported [23].
The standard yeast selection medium YPDS (Yeast Extract Peptone Dextrose with Sorbitol) comprised 1% yeast extract, 2% peptone, 2% dextrose (glucose), 1 M sorbitol, 2% agar (all w/v, Invitrogen, Carlsbad, CA, USA). The standard yeast expression media BMGY/BMMY (Buffered Glycerol-complex Medium/Buffered Methanol-complex Medium, both from Invitrogen) comprised 1% yeast extract, 2% peptone, 100 mM potassium phosphate, pH 6.0, 1.34% YNB, 4 × 10−5% biotin (all w/v), 1% glycerol or 0.5% methanol (both v/v).
Cloning, expression and purification of recombinant enzyme
The genomic DNA encoding the laccase of auxiliary family 1 [9] (http://www.cazy.org/) from Zymoseptoria tritici IPO323 (GenBank accession XP003852363) was synthesized by Invitrogen (Carlsbad, CA, USA). The forward GACATTTCGAAACGATGCGGTTAC and reverse primer GATTGTCTAGATCAGACTCCGGAATC were used to amplify this gene fragment that encodes the mature peptide of the enzyme lacking the native signal peptide (amino acid residues 46–723, hereafter designated as ZtrLac1A) using Turbo DNA polymerase and a PCR protocol involving preheating at 95 °C (30 s), followed by 16 cycles of denaturation at 95 °C (30 s), annealing at 55 °C (1 min) and extension at 68 °C (1 min). The PCR amplicon (2197 bp) was cloned within the BstBI and XbaI restriction sites of pPICZαA vector (Invitrogen, Carlsbad, CA, USA) using standard molecular biology protocols. The resulting recombinant pPICZαA-Ztrlac1A plasmid was transformed into Escherichia coli DH5α and clones were selected on low salt LB agar plates supplemented with 25 μg mL− 1 Zeocin. Sequencing was performed using the 5′-AOX promoter 3′-AOX terminator universal primers and the internal primers 5′-GGGTTTGAATTATGAGGATCCG-3′ and 3′-TTGACTTGCCAGTAGAGGGTG-5′).
The recombinant plasmid was linearized using PmeI and transformed by electroporation into Pichia pastoris X-33 cells according to the manufacturer’s protocol. Transformants were selected on YPDS plates supplemented with 100 μg mL− 1 Zeocin according to the manufacturer’s recommendation (EasySelect™ Pichia Expression Kit, Invitrogen) after incubation at 30 °C for 3–5 days. Single transformants were re-streaked on new plates and used to inoculate 5 ml buffered complex medium BMGY. The best secreting transformants were selected based on the SDS-PAGE analysis and activity measurements using ABTS as substrate.
An overnight culture (25 mL, BMGY medium, 30 °C) was grown to OD600 = 5 prior to inoculation into 0.9 L of the same medium and propagated overnight to OD600 ≈ 7. Thereafter, cells were harvested by centrifugation (7000 g, 30 min, 4 °C), re-suspended in BMMY to OD600 ≈ 1, and re-incubated at 30 °C. The culture was supplemented with methanol (0.5% v/v) every 24 h prior to harvesting after 72 h of induction. Culture supernatants were recovered by centrifugation (7500 g, 30 min, 4 °C), pooled, supplemented with (NH4)2SO4 to 0.5 M and incubated for 18–20 h, followed by centrifugation (8000 g, 20 min, 4 °C) and filtration (0.5 μm) prior loading onto a 40 ml β-cyclodextrin-Sepharose affinity column and purification as previously described [24]. The elution fractions containing ZtrLac1A were analyzed using SDS-PAGE and activity measurements according to the protocol described below. Fractions displaying high purity and activity were pooled and concentrated using Amicon filters (MWCO, 30 kDa, Millipore). The concentrated protein loaded onto a HiLoad 36/60 Superdex gel filtration column (GE Healthcare, Uppsala, Sweden) and eluted with 20 mM Na acetate, 150 mM NaCl, pH 5.5 at 0.9 ml min− 1. The fractions with highest purity and activity were pooled, concentrated and the protein concentration was determined by measuring the absorbance at 280 nm (A280) using the theoretical extinction coefficient ε280 = 79074 M− 1.cm− 1 Expasy (http://web.expasy.org/protparam). All Purification steps were performed using an ÄKTA purifier chromatograph (GE Healthcare) at 7 °C. N-Glycosylation and O-glycosylation sites were predicted using NetNGyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/), and NetOGlyc (http://www.cbs.dtu.dk/services/NetNGlyc/), respectively. To examine N-glycosylation, a 9 μl of supernatant was initially denatured at 100 °C (10 min) and furthermore was used in EndoH treatment at 37 °C for 90 min according to the manufacturer’s instructions (New England Biolabs) and finally analyzed using SDS-PAGE.
Isothermal titration calorimetry
The isothermal titration calorimetry (ITC) experiments were performed using an iTC200 instrument (GE healthcare, Northampton, MA, USA). ZtrLac1A (145 μM) dialyzed against 20 mM NaOAc, pH 5.5 was titrated with 2.1 mM β-cyclodextrin (β-CD) dissolved in the same buffer at 25 °C with an initial injection of 0.4 μL followed by 15 injections of 2 μL. A control titration into the dialysis buffer was used to compensate the heat of dilution. A one-binding site model was fit to the ITC data to determine the equilibrium association constant (Ka), the molar binding enthalpy (ΔH) and the stoichiometry of binding (No) using the ITC analysis plug in ORIGIN software provided with the instrument.
Biochemical characterization
The kinetic parameters of ZtrLac1A (Km and kcat) were determined by monitoring the increase in absorbance of oxidized ABTS (A405, ε405 = 36.8 mM− 1 cm− 1) [25] in 96-well microtiter plates using a PowerWave XS plate reader (BioTeK, Winooski, VT, USA) using eight different concentrations (0.032–4 mM) in 40 mM sodium acetate buffer pH 4.2 at 25 °C. The ZtrLac1A activity as function of pH was measured for 10 min at 25 °C in the pH range 2.2–8.0, using ABTS, as substrate in McIlvaine buffer.
The laccase activity was also determined using 2,6-DMP (A469, ε469 = 19.6 mM− 1 cm− 1) and syringaldazine (A525, ε525 = 65.0 mM− 1.cm− 1) in the range of 0.4–8 mM and 0.006–0.11 mM, respectively. The reactions were initiated by the addition of the enzyme to a final concentration of 0.26 μM to pre-temperated mixtures containing substrate and buffer to a final volume of 250 μL. The experiments were carried out in triplicates and the kinetic parameters were determined by fitting the Michaelis-Menten equation to the initial rate data at each substrate concentration using ORIGIN 8.0 (OriginLab, Northampton, MA). An additional activity assay was also carried out using the phenolic substrates coniferyl aldehyde, p-coumaric, caffeic acids, vanillin, 3,5 dimethoxybenzoic acid and 3,4 dihydroxybenzoic acid in 2 mM concentrations. All the reactions were performed in 50 mM NaAc pH 5.2 in 0.5 ml reaction volume for 30 and 90 min. The structures of these substrates and their analysis are described in Additional file 1: Figure S2a–e.
Electrochemical measurements and electrode modification
The redox potential of the T1 site for ZtrLac1A was determined using modified graphite electrode (GE) in the presence and absence of a redox mediator. Electrodes were prepared as previously described for bioelectrochemical studies on multicopper oxidase (MCO) [26]. The spectrographic low-density graphite rods (Ø 3.05 mm, Sigma) were polished by fine emery paper (Turfbak Durite, P1200), thoroughly rinsed with Milli-Q water and dried at room temperature. The GE/Os/ ZtrLac1A (0.16 mM) modified electrodes were prepared by transferring Os-polymer (5 μL, 5 mg mL− 1) and freshly prepared poly(ethylene glycol) diglycidyl ether solution (PEGDGE, 2 μL, 10 mg/mL water) to the top of the GE and subsequent incubation for 10 min, followed by adding the enzyme solution (12.6 mg/mL). Thereafter, the modified electrodes were dried at room temperature prior to incubation overnight at 4 °C to complete the cross-linking. Furthermore, in the direct electron transfer approach (in absence of a redox mediator), the same immobilization procedure was also applied but in the absence of the Os-polymer and cross-linking agent. To begin with, the enzyme-modified electrodes were press-fitted into a Teflon holder and rinsed thoroughly with acetate buffer to remove any weakly bound material. All electrode modifications were prepared in triplicate.
A three-electrode cell was used with an Ag|AgCl KClsat (E = 0.199 V vs. SHE, all redox potentials are reported vs. SHE) as reference electrode and a Pt plate as counter electrode. The laccase modified electrodes were used as working electrode. Cyclic voltammetry measurements were performed in an 0.1 M acetate buffer at pH 4.0 also containing 0.1 M NaClO4 at room temperature. Prior to the measurements in the absence of O2, N2 gas was bubbled through the measuring solution at least for 20 min to maintain O2-free conditions. Oxygen saturated conditions were achieved by purging pure O2 in the buffer solution for at least 20 min. All cyclic voltammetry measurements were conducted using a potentiostat Metrohm Autolab Potentiostats/Galvanostats (Model PGSTAT128N, Metrohm Autolab B.V., Utrecht, The Netherlands) equipped with Nova 2.1 as software.
Phylogenetic analysis and homology modelling
Most of the ascomycete laccases (about 90%) assigned into AA1_3 in the CAZy database were retrieved. In addition, the catalytic module of ZtrLac1A was also used as a query in a BLASTp search (https://www.ncbi.nlm.nih.gov/BLAST/) against the non-redundant protein sequence database [27] to retrieve additional basidiomycete sequences within an E-value < 5 × 10− 71, sequence identity > 31% and a minimum sequence coverage of 88%. In total 221 sequences from ascomycete and basidiomycetes laccase catalytic modules (AA1) were aligned using MAFFT software standard settings (mafft.cbrc.jp/alignment/). The phylogenic tree, calculated using neighbor joining method with bootstrap value of 1000 in MAFFT server and it was visualized by DENDROSCOPE [28].
A homology model for ZtrLac1A was generated using the laccase structure from Melanocarpus albomyces (PDB code 1GW0, 44% sequence identity) as a template and the Schrödinger package. The template enzyme represents the closest structurally and biochemically characterized (Table 1) homologue of ZtrLac1A.
Results
Heterologous expression of the modular laccase from Zymoseptoria tritici (ZtrLac1A)
The non-codon optimized synthetic gene encoding the modular laccase from Zymoseptoria tritici (ZtrLac1A) (Additional file 1: Figure S3a), was cloned into Pichia pastoris. The recombinant protein was expressed and purified using β-cyclodextrin-Sepharose affinity chromatography, confirming the binding functionality of the CBM20. The purification was polished with an additional gel filtration step and the yield of pure protein was about 1.5 mg L− 1 culture and a specific activity of 45.5 U mg− 1. ZtrLac1A migrated as smeary bands with an apparent molecular mass of 93 kDa compared to the theoretically calculated value (79 kDa). The larger size is consistent with O- and/or N-glycosylation, which are typically observed in recombinant proteins produced in P. pastoris [30]. Among seven putative N-glycosylation sites in the catalytic module, only three exhibit high score (> 0.6), Asn 209, Asn252 and Asn366. The putative O-glycosylation sites were predicted in serine/threonine rich linker regions (Additional file 1: Figure S3a) [31]. Endo-H treatment of the recombinant enzyme resulted in a reduction of about 15 kDa in molecular mass estimated from the migration shift in SDS-PAGE analysis (Additional file 1: Figure S3b) suggesting the protein is mainly decorated with N-glycans.
ZtrLac1A is a functional laccase
The activity of ZtrLac1A was examined using the synthetic mediator ABTS at pH 4.2, whereas activity on syringaldazine and 2,6-DMP was assayed at pH 6.0. Notably, ZtrLac1A is mainly active on ABTS, compared to a negligible activity towards 2,6-DMP and no activity towards syringaldazine, which is different from the closest related ascomycete laccases from Melanocarpus albomyces laccase (MalLac1A) (Table 1).
Isothermal titration calorimetry (ITC)
The binding of ZtrLac1A to the starch mimics β-CD, a typical model substrate used to demonstrate the functionality of starch-binding proteins [32], was quantified using ITC (Fig. 2). The data revealed that ZtrLac1A binds to β-CD with an association constant of Ka = (3.07 ± 0.24) × 104 M− 1 (Kd = 32.6 ± 2.4 μM, ΔG = − 6.17 kcal mol− 1). The binding was largely driven by a favorable enthalpy (ΔH = − 7.15 ± 0.19 kcal mol− 1), which is compensated by an unfavorable change in entropy (−TΔS = 1.03 kcal mol− 1). The fitted stoichiometry of No = 1.5 ± 0.1 is lower, but consistent with the presence of two β-CD binding sites in canonical CBM20 modules [32], which is likely to be due to residual partial occupancy from the β-CD elution during purification.

Isothermal titration calorimetry of the binding of ZtrLac1A to β-CD, carried out at 25 °C at pH 5.5. a Binding thermogram and b The normalized integrated heat response (black squares) plotted against the molar ratio of injected ligand to the protein concentration in the cell. The fit of a one-site binding model (black line) to the binding isotherm
Electrochemical characterization
In order to study the electrocatalytic behavior of ZtrLac1A, modified graphite electrode (GE) cyclic voltammograms (CVs) were collected in absence (black curve) and presence (red curve) of O2 in 0.1 M acetate buffer at pH 4.0, 0.1 M NaClO4 (Fig. 3a). It was not possible to define the onset potential for the electroreduction of O2 from these CVs, while this was clearly displayed in the subtracted curve at 0.569 V vs. SHE (Fig. 3). A second electroreduction process was observed at approximately 0.2 V and attributed to direct non-enzymatic oxygen reduction at the GE surface. Unmodified GE studied at the same conditions showed only electro-reduction at 0.2 V as shown in (Additional file 1: Figure S4). The peaks observed with E°′ 0.408 V vs. SHE (Fig. 3a) are attributed to quinone moieties at the GE as previously reported [33, 34]. The ZtrLac1A modified GE electrodes do not show O2 electroreduction at this potential implying that there is no electrical communication between these quinone moieties and the enzyme.

a Cyclic Voltammograms (CVs) recorded with ZtrLac1A modified low-density GE in N2 (black line) and in O2 (red line); (inset Fig. 3a) subtracted curve in order to determine the onset of O2 reduction. b CVs recorded with GE/Os-polymer/ZtrLac1A under N2 (black line) and in O2 (red line) conditions. (Inset Fig. b) subtracted curve in order to determine the onset of O2 reduction. Conditions: 0.1 M acetate buffer at pH 4.0 in 0.1 M NaClO4; scan rate 10 mVs− 1
Furthermore, to confirm the results, ZtrLac1A was co-immobilized with an Os-redox polymer and polyethylene glycol diglycidyl ether (PEGDGE) (GE/Os/ZtrLac1A) using previously reported methods [35, 36]. An Os- polymer with (E°′ = 0.549 V vs. SHE) was used as a surface immobilized redox mediator [37] to facilitates electron transfer between electrode surface and the T1 site of the enzyme. This mediated electron transfer is thermodynamically favorable if the E°‘of the polymer is more negative than E°’ of the T1 site. A polymer with E°′ close to that of the T1 site is desirable. This together with the highly flexible backbone and highly cationic nature of the polymer facilitate enzyme immobilization and electron transfer between the Os2+/3+ containing complexes, the enzyme and electrode. The polypyrrole side chains present reactive –NH2 groups, which are used in combination with PEGDGE to stabilize the immobilization through covalently links between the enzyme and polymer. This results in a 3D-hydrogel incorporating very high amounts of both immobilized enzyme molecules as well as mediating functionalities onto the electrode surface with easy access for substrates and products to diffuse to/from the enzyme active site. The electrochemical characterization of ZtrLac1A entailed the determination of the onset potential for the oxygen electro-reduction (EO2), which is close to the formal potential of the T1 site (ET1) [38]. The onset potential was estimated by physically adsorbing the enzyme onto graphite electrodes (GE) with or without an Os-polymer of E°′ 0.549 V vs. Standard hydrogen electrode (SHE) previously characterized [37].
The Os-polymer facilitates transfer of electrons between the electrode and enzyme in a process known as mediated electron transfer (MET). CVs corresponding to GE/Os/ZtrLac1A are shown in Fig. 3b with the subtracted curves, where the peaks observed at 0.532 V vs. SHE are attributed to the Os-polymer. Two electrocatalytic processes were observed in the presence of O2 with the first having its onset potential at 0.660 V vs. SHE, which is attributed to the MET between the Os-polymer and ZtrLac1A. The second process starting from 0.4 V vs. SHE is attributed to the direct electroreduction of O2 in the surface of the GE as previously discussed.
Phylogenetic and sequence analysis
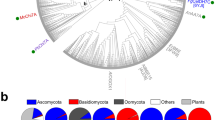
The aligned sequences were from the ascomycetes laccases retrieved from the CAZy database and the basidiomycetes sequences from a BLASTp search of the non-redundant protein sequences database using the catalytic module of ZtrLac1A (556 residues) as a query. This analysis revealed that the closest orthologues to ZtrLac1A are laccases from the plant pathogens Zymoseptoria brevis (ZbrLac1A) and Sphaerulina musiva (SmuLac1A) sharing 98 and 69% sequence identity, respectively. In total, 221 sequences (490–650 amino acid residues, 55–75 kDa), including ascomycete and basidiomycetes laccases, were included in the alignment and phylogenetic tree.
ZtrLac1A together with eight closest homologues segregate into a single branch, which is hereafter designated as the Septoria cluster in the phylogenetic analysis. Remarkably, six of these putative laccases share the same modular organization as ZtrLac1A (Additional file 1: Figure S3a) with variations in the length of linker (12–17 amino acid residues) separating the binding and catalytic modules (Additional file 1: Table S1). The Septoria cluster (bold blue in Fig. 4) is adjacent to the two closest structurally characterized ascomycete laccases, Melanocarpus albomyces (MalLac1A) and Thielavia arenaria (TarLac1A), displaying 44% sequence identity to ZtrLac1A (in brown). Two other structurally characterized ascomycete laccases from Botyris aclada (BacLac1A) and Aspergillus niger (AniLac1A) [39] that share 41 and 36% sequence identity to ZtrLac1A, respectively, segregate in two distant clades. The basidiomycetes orthologues populate a distinct clade, which includes some intermediate ascomycete sequences from A. niger and Fusarium oxysporum (Fig. 4).

Phylogenetic analysis of ZtrLac1A and related fungal laccases. The phylogram shows 221 orthologues to the catalytic module of ZtrLac1A, all assigned into AA1 according to the CAZy classification. The Septoria cluster is highlighted in blue and the two sequences from the Zymoseptoria tritici species are in bold, whereas basidiomycetes sequences are in black. The only four structurally characterized ascomycete laccases are in brown. The tree illustrates the segregation of Septoria in a distinct clade of the phylogenetic tree consistent with the unique biochemical profile and the prevalent occurrence with a CBM20 module as opposed to other enzymes
Distinctive structural features in ZtrLac1A compared to other fungal laccases
To analyse the structural elements responsible for segregation of the ZtrLac1A in a distinct cluster, we compared these enzymes to related sequences of AA1_3 in CAZy. To assess the functional relevance of these differences, we generated homology models of ZtrLac1A based on the closest structurally characterized ascomycete laccase from M. albomyces (MalLac1A) as a template (44% identity to ZtrLac1A). A good quality model was obtained as judged by LGscore and MaxSub qualitative values of 6.18 and 0.24 calculated via the ProQ online server [40], respectively. Overall, the model was similar to MalLac1A as reflected by the root mean square deviation (RMSD) of 0.78 Å for the superimposition of 528 Cα backbone atoms out of 556 between these enzymes, which also shared three disulphide bridges that stabilize their structures. In ZtrLac1A, the disulfide bridges between Cys303-Cys340, Cys118-Cys544, and Cys10-Cys19 at the N-terminus stabilize the fold analogous to counterparts in the TarLac1A and AniLac1A structures.
The sequence alignment between ZtrLac1A and the characterized laccases revealed differences in three functionally relevant regions including loops surrounding the active site, C-terminus and copper 1 (T1) active center.
The substrate-binding site in fungal laccases is defined by four loops that flank the T1 site [39]. Ascomycete laccases have elongated loops in this region compared to basidiomycetes laccases. Notably, Loop A (N373 − L384) in ZtrLac1A is further elongated with two, four and six residues compared to the corresponding loops in the known structural homologues AniLac1A, MalLac1A, TarLac1A, and BacLac1A, respectively (Fig. 5a). Significant sequence substitutions are also observed in this loop, e.g. the T368, N363 and V387 in MalLac1A (and TarLac1A), BacLac1A and AniLac1A laccases are substituted with the K381 in ZtrLac1A (Fig. 5a). Similarly, Loop B comprising T435 − N444 in ZtrLac1A, is also elongated with up to six residues compared to TarLac1A, MalLac1A, BacLac1A as well as AniLac1A possessing the shortest B loop. Notably, a proline (P423) in MalLac1A and TarLac1A, is substituted with glutamine (Q437) in ZtrLac1A (Additional file 1: Figure S5).

Superimposition of modeled ZtrLac1A with the structures from the ascomycetes M. albomyces (MalLac1A, 1GW0), T. arenaria (TarLac1A, 3PPS), Botyris aclada (BacLac1A, 3SQR) and A. niger (AniLac1A, 5LM8). a The loop A flanking the T1 substrate binding site in ZtrLac1A (dark blue) is variable with respect to size and sequence from the structurally characterized MalLac1A (light brown), TarLac1A (red), BacLac1A (Cyan) as well as AniLac1A (green). b Superposition of C-termini of ZtrLac1A, MalLac1A, TarLac1A, BacLac1A and AniLac1A. The Cu 1 and Cu 2–3 are shown as red spheres. Structures were rendered using PYMOL v1.8 software (Schrödinger, LLC, Palo Alto, CA). The sequence alignment of these functionally relevant structural elements is shown and divergent segments and amino acid substitutions that involve large changes in chemistry or length of sidechains are highlighted in grey shades
ZtrLac1A also displays important difference at the C-terminus, with three proline residues in the structurally characterized laccases, being substituted to leucine, glutamine or lysine (Fig. 5b). Mutation of the leucine at this position to methionine was shown to reduce the activity of BalLac1A highlighting the functional importance of C-terminus [41].
Discussion
The first starch binding modular laccase from fungal pathogens that use apoplastic infection strategy
Carbohydrate active enzymes (CAZymes) are frequently modular featuring one or more catalytic modules (CMs) appended to non-catalytic auxiliary modules. The most common non-catalytic modules are ancillary CBMs, which potentiate the deconstruction of insoluble polysaccharides by mediating substrate targeting and prolonged contact of cognate enzymes [42]. Presence of CBMs is also observed in oxidoreductases targeting complex polysaccharides [43]. In this study, we report the properties of the first modular laccase possessing a starch-binding CBM20. This enzyme stems from the major wheat pathogen Z. tritici causing massive crops yield losses globally [17, 18]. Z. tritici has a “stealth” pathogenesis, with an apoplastic and symptomless initial growth within the plant cell wall. During later stages, loss of plant cell integrity, leakage of nutrients to the intercellular space concomitant with acceleration of fungal growth and finally necrotic lesions on leaves are typically observed [19]. Despite the significant reduction in CAZymes and CBMs [44], the genome of Z. tritici encodes at least 24 putative starch degrading enzymes including α-glucosidases (8 of GH31 and 1 of GH133), α-amylases (14 sequences from GH13) and a glucoamylase (GH15) (http://www.cazy.org) [45]. This genomic expansion with putative starch targeting enzymes is also reflected by the presence of seven encoded proteins possessing putative starch binding domains including four assigned into CBM20. Notably, large changes in chloroplasts are observed during infection, especially during the later stages, which revealed an expansion of chloroplasts before cell collapse [46]. The chloroplasts are the sites of starch synthesis in the leaves [46], and interestingly a recent transcriptional analysis revealed a substantial upregulation of α-amylases and oxidoreductases in the taxonomically related pine pathogen Dothistroma septosporum also during the later stage of infection [47]. This fungus possesses a homologue of the Z. tritici laccase characterized in the present study (Additional file 1: Figure S3, Additional file 1: Table S1). In fact, all the seven modular laccases possessing a CBM20 stem from taxonomically related pathogens from the order Capnodiales. Taken altogether, the evolution of starch binding laccases in this group of pathogens and the histological and transcriptional response seems to highlight an important role of starch targeting enzymes in the necrotic stage of infection. Our data provide compelling evidence for the functionality of the CBM20, which possesses moderate affinity (Kd 33 μM) typical of counterparts from amylolytic enzymes. Therefore, ZtrLac1A is likely to be efficiently targeted to starch granules, but the role of the laccase and the substrates it oxidizes at the starch granule surface in vivo remain intriguing and merits further studies.
ZtrLac1A displays a unique activity profile and sequence signatures compared to ascomycete characterized laccases
The kinetic data of ZtrLac1A on the negatively charged redox mediator ABTS verified its laccases functionality (Table 1). Interestingly, the Km of this enzyme on ABTS was 60% lower than that measured for the closest related ascomycete laccases MalLac1A (Table 1). While, MalLac1A displays an increasing affinity and efficiency on the more apolar phenolic mediators DMP and syringaldazine compared to ABTS [29], ZtrLac1A was almost inactive on these substrates (Table 1). This sequence signature is also consistent with changes in the sequence and length of loops flanking the T1 binding site, where the substrate binding and oxidation occurs as compared to MalLac1A and other currently characterized ascomycete laccases (Fig. 5, Additional file 1: Figure S5). Notably, the loops flanking the active site of ZtrLac1A, exhibit a lower content of negatively charged and apolar residues compared to structurally characterized laccases. These changes, together with the elongation of loop A with four amino acid residues including a lysine (Fig. 5a), are consistent with a more positive electrostatics and occlusion of the active site in ZtrLac1A. These substitutions may provide a rationale for better affinity of the negatively charged ABTS and the lack of activity on the two other less polar mediators. The C-terminal loop of laccases acts as a plug that occludes the T2 and T3 sites, where the second substrate O2 binds [29]. A single mutation of the C-terminal residue (L559A) and the deletion of the four C-terminal residues of MalLac1A caused a severe activity drop of the enzyme [29]. We have also observed important substitutions in this C-terminal segment making it more flexible compared to characterized counterpart due to the loss of three closely located proline residues (Fig. 5b).
Another interesting difference is a conserved residue close to the Cu ligands at the T1 site (Additional file 1: Figure S6). This residue is either a phenylalanine as in ZtrLac1A, or a leucine as in characterized ascomycete laccases (Additional file 1: Figure S6), but methionines are also observed in a few non-characterized ascomycete laccases. Site-directed mutagenesis of Cu ligands have shown large effects on the redox potential [39, 48]. According to the current classification, laccases are arranged in three groups: low (340–490 mV), middle (470–710 mV) and high potential (730–780 mV) [49]. The difference in the T1 potentials depends strongly on the axial Cu-ligands of the T1 site being methionine for low potential, leucine for middle and phenylalanine for high potentials [49].
The bioelectrochemical O2 reduction by ZtrLac1A immobilized on GE electrodes gives an estimate of the potential of T1, being 0.569 V vs. SHE when the electrons are transferred directly from the electrode to the T1 site of the enzyme. The successfully mediated electroreduction observed for GE/Os/ZtrLac1A is an indication of the closeness of the E0’ of T1 to the one of the Os-polymer used (0.55 V). The assignment of the onset to be from the T1 site is based on previously published results reporting the electro-reduction of O2 at carbon modified electrodes with “blue” MCO [50,51,52]. Subsequently, electron transfer proceeds internally through the T2/T3 multicopper site, where the electroreduction of O2 takes place.
ZtrLac1A is thus classified as a middle redox potential accordingly, which is in agreement with previous values reported for the few other ascomycete characterized laccases [48].
Conclusions
ZtrLac1A displays an atypical substrate preference, but shows a typical redox potential range compared to ascomycetes characterized counterparts. This first demonstration of activity of a modular laccase with a starch binding module offers an attractive possibility for reversible immobilization of laccases on cheap material like starch or cellulose, and allows targeting of these enzymes without using classical covalent immobilization techniques that typically reduce enzymatic activity. Finally, further studies are needed to get insight into the role of this enzyme in the pathogenic life cycle of fungi from the order Capnodiales and to identify physiologically relevant substrates.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- 2,6-DMP:
-
2,6-dimethoxyphenol
- ABTS:
-
2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)
- AniLac1A:
-
Aspergillus niger laccase
- BacLac1A:
-
Botyris aclada laccase
- CBM:
-
Carbohydrate binding module
- CMs:
-
Catalytic modules
- CV:
-
Cyclic voltammograms
- GE:
-
Graphite electrode
- MalLac1A:
-
Melanocarpus albomyces laccase
- MCOs:
-
Multicopper oxidases
- SHE:
-
Standard hydrogen electrode
- STB:
-
Septoria tritici blotch
- TarLac1A:
-
Thielavia arenaria laccase
- ZtrLac1A:
-
Zymoseptoria tritici laccase
- β-CD:
-
β-cyclodextrin
References
Solomon EL, Sundaram UM, Machonkin TE. Multicopper oxidases and oxygenases. Chem Rev. 1996;96:2563–606.
Giardina P, Faraco V, Pezzella C, Piscitelli A, Vanhulle S, Sannia G. Laccases: a never-ending story. Cell Mol Life Sci. 2010;67:369–85.
Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Copper active sites in biology. Chem Rev. 2014;114:3659–853.
Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Oxygen binding, activation, and reduction to water by copper enzymes. Angew Chem Int Ed Engl. 2001;40:4570–90.
Kamitaka Y, Tsujimura S, Kataoka K, Sakurai T, Ikeda T, Kano K. Effects of axial ligand mutation of the type I copper site in bilirubin oxidase on direct electron transfer-type bioelectrocatalytic reduction of dioxygen. J Electroanalytical Chem. 2007;601:119–24.
Reinhammar BRM. Oxidation-reduction potentials of the electron acceptors in laccases and stellacyanin. Biochim Biophys Acta. 1972;275:245–59.
Rodjers CJ, Blandford CF, Giddens SR, Skamnioti P, Armstrong FA, Gurr SJ. Designer laccases: a vogue for high-potential fungal enzymes? Trends Biothechnol. 2010;28:63–72.
Claus H. Laccases: structure, reactions, distribution. Micron. 2004;35:93–6.
Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6:41.
Kiiskinen LL, Viikari L, Kruus L. Purification and characterisation of a novel laccase from the ascomycete Melanocarpus albomyces. Appl Microbiol Biotechnol. 2002;59:198–204.
Baldrian P. Fungal laccases - occurrence and properties. FEMS Microbiol. 2006;30:215–42.
Slomczynski D, Nakas JP, Tanenbaum SW. Production and characterization of laccase from Botrytis cinerea. Appl Environ Microbial. 1995;61:907–12.
Iyer G, Chattoo BB. Purification and characterization of laccase from the rice blast fungus, Magnaporthe grisea. FEMS Microbiol Lett. 2003;227:121–6.
Canero Cordoba D, Roncero MIG. Functional analyses of laccase genes from Fusarium oxysporum. Amercian phytopathol. Soci. 2007;98:509–18.
O’Driscoll A, Kildea S, Doohan F, Spink J, Mullins E. The wheat-Septoria conflict: a new front opening up? Trends Plant Sci. 2014;19:602–10.
Torriani SFF, Melichar JPE, Mills C, Pain N, Sierotzki H, Courbot M. Zymoseptoria tritici: a major threat to wheat production, integrated approaches to control. Fungal Genet Biol. 2015;79:8–12.
Fones H, Gurr S. The impact of Septoria tritici blotch disease on wheat: an EU perspective. Fungal Genetic Biol. 2015;79:3–7.
Jørgensen LN, Hovmøller MS, Hansen JG, Lassen P, Clark B, Bayles R, B. Rodemmann, Flath K, Jahn M, Goral T, et al. IPM strategies and their dilemmas including an introduction to www.eurowheat.org. J Integr Agric 2014;13:265–281.
Dean R, Van Kan JA, Pretorious ZA, Hammond-Kosack KE, Di Pietro A, Spanu PD, Rud JJ, Dickman M, Kahmann R, Elis J, Foster GD. The top 10 fungal pathogens in molecular plant pathology. Mol Plant Pathol. 2012;13:414–30.
Windels CE. Economic and social impacts of Fusarium head blight: changing farms and rural communities in the northern Great Plains. Phytopathology. 2000;90:17–21.
Howard RJ, Valent B. Breaking and entering: host penetration by the fungal rice blast pathogen Magnaporthe grisea. Annu Rev Microbiol. 1996;50:491–512.
Goodvin SB, MBarek SB, Dhillon B, et al. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals Dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011. https://doi.org/10.1371/journal.pgen.1002070.
Mano N, Kim HH, ZhangY, Heller A. An oxygen cathode operating in a physiological solution. J Am Chem Soc. 2002;124:6480–6.
Nekiunaite L, Isaksen T, Vaaje-Kolstad G, Abou Hachem M. Fungal lytic polysaccharide monooxygenases bind starch and β-cyclodextrin similarly to amylolytic hydrolases. FEBS Lett. 2016;590:2737–47.
Niku-Paavola ML, Karhunen E, Salola P, Raunio V. Ligninolytic enzymes of the white-rot fungus Phlebia radiate. Biochem J. 1988;254:877–84.
Shleev S, Jarosz-Wilkolazka A, Khalunina A, Morozova O, Yaropolov A, Ruzgas T, Gorton L. Direct heterogeneous electron transfer reactions of laccases from different origins on carbon electrodes. Bioelectrochemistry. 2005;67:115–24.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids. 1997;25:3389–402.
Huson DH, Scornavacca C. Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst Biol. 2012;61:1061–7.
Andberg M, Hakulinen N, Auer S, Saloheimo M, Koivula A, Rouvinen J, Kruus K. Essential role of the C-terminus in Melanocarpus albomyces laccase for enzyme production catalytic properties and structure. FEBS J. 2009;276:6285–300.
Borodina I, Jensen BM, Wagner T, Abou Hachem M, Sondergaard I, Poulsen LK. Expression of enzymatically inactive wasp venom phospholipase A1 in Pichia pastoris. PLoS One. 2011;6:e21267.
Jeoh T, Michener W, Himmel ME, Decker SR, Adney WS. Implications of cellobiohydrolase glycosylation for use in biomass conversion. Biotechnol Biofuels. 2008;1:10.
Christiansen C, Abou Hachem M, Janecek S, Viksø-Nielsen A, Blennow A, Svensson B. The carbohydrate-binding module family 20-diversity, structure, and function. FEBS J. 2009;276:5006–29.
Swain GM. Handbook of electrochemistry, ed. C G Zoski Elsevier Amsterdam. 2007;111-V.
McCreery RL. Advanced carbon electrode materials for molecular electrochemistry. Chem Rev. 2008;108:2646–87.
Heller A. Electrical wiring of redox enzymes. Acc Chem. 1990;23:128–34.
Zafar MN, Tasca F, Boland S, Kujawa M, Patel I, Peterbauer CK, Leech D, Gorton L. Wiring of pyranose dehydrogenase with osmium polymers of different redox potentials. Bioelectrochemistry. 2010;8038–42.
Kavanagh P, Jenkins P, Leech D. Electroreduction of O2 at a mediated Melanocarpus albomyces laccase cathode in a physiological buffer. Electrochem Commun. 2008;10(7):970–2.
Dos Santos L, Climent V, Blandford CF, Armstrong FA. Mechanistic studies of the ‘blue’ cu enzyme, bilirubin oxidase, as a highly efficient electrocatalyst for the oxygen reduction reaction. Phys Chem Chem Phys. 2010;12:13962–74.
Ferraroni M, Westphal AH, Borsari M, Tamayo-Ramos JA, Briganti F, de Graaff LH, van Berkel WJH. Structure and function of Aspergillus niger laccase McoG. Biocatalysis. 2017;3(1):16.
Wallner B, Elofsson A. Protein Sci. 2003;12:1073–86.
Osipov E, Polyakov K, Kittl R, Shleev S, Dorovatovsky TT, Hann S, Ludwig R, Popov V. Effect of the L499M mutation of the ascomycetous Botrytis aclada laccase on redox potential and catalytic properties. Acta Crystallogr D Biol Crystallogr. 2014;70:2913–23.
Gilbert HJ, Knox JP, Boraston AB. Advances in understanding the molecular basis of plant cell wall polysaccharide recognition by carbohydrate-binding modules. Curr Opin Struct Biol. 2013;23:669–77.
Nekiunaite L, Arntzen M, Svensson B, Vaaje-Kolstad G, Abou Hachem M. Lytic polysaccharide monooxygenases and other oxidative enzymes are abundantly secreted by Aspergillus nidulans grown on different starches. Biotechnol Biofuel. 2016;9:187.
do Amaral AM, Antoniw J, Rudd H-KKE. Defining the predicted protein Secretome of the fungal wheat leaf pathogen Mycosphaerella graminicola. PLoS One. 2012;7:e49904.
Kema GHJ, Yu DZ, Rijkenberg FHJ, Shaw MW, Baayen RP. Histology of the pathogenesis of Mycosphaerella graminicola in wheat. Phytopathology. 1996;86:777–86.
Zeeman SC, Delatte T, Messerli G, Umhang M, Stettler M, Mettler T, Streb S, Reinhold H, Kötting O. Starch breakdown: recent discoveries suggest distinct pathways and novel mechanisms. Funct Plant Biol. 2007;34:465–73.
Bradshaw RE, Guo YD, Sim AD, Kabir MS, Chettri P, Ozturk IK, Hunziker L, Ganley RJ, Cox MP. Genome-wide gene expression dynamics of the fungal pathogen Dothistroma septosporum throughout its infection cycle of the gymnosperm host Pinus radiate. Mol Plant Pathol. 2016;17:210–24.
Durão P, Chen Z, Silva CS, Soares CM, Pereira MM, Todorovic S, Hildebrandt P, Bento I, Lindley PF, Martins LO. Proximal mutations at the type 1 copper site of CotA laccase: spectroscopic, redox, kinetic and structural characterization of I494A and L386A mutants. Biochem J. 2008;412:339–46.
Shleev S, Tkac J, Christenson A, Ruzgas T, Yaropolov AI, Whittaker JW, Gorton L. Direct electron transfer between copper-containing proteins and electrodes. Biosens Bioelectron. 2005;20:2517–54.
Berezin IV, Bogdanovskaya VA, Varfolomeev SD, Tarasevich MR, Yaropolov AI. Bioelectrocatalysis. Equilibrium oxygen potential in the presence of laccase. Dokl Akad Nauk SSSR. 1978;240:615–8.
Yaropolov AI, Kharybin AN, Emnéus J, Marko-Varga G, Gorton L. Electrochemical properties of some copper-containing oxidases. Bioelectrochem Bioenerg. 1996;40:49–57.
Christenson A, Dimcheva N, Ferapontova EE, Gorton L, Ruzgas T, Stoica L, Shleev S, Yaropolov AI, Haltrich D, Thorneley RNF, Aust SD. Direct electron transfer between ligninolytic redox enzymes and electrodes. Electroanalysis. 2004;16:1074–92.
Acknowledgments
We are grateful to Prof. Dónal Leech and Dr. Peter Ó Conghaile from Biomolecular Electronics Research Laboratory; National University of Ireland Galway (Galway, Ireland) for providing us with osmium polymer.
Funding
This study was supported by a Novo Nordisk foundation grant (NNF12OC0000769) within the “Biotechnology-based Synthesis and Production” programme to MAH, and a H.C. Ørsted-Marie Curie Actions COFUND Post-doc grant and Novo Nordisk foundation (NNF17OC0025642) within Biotechnology-based Synthesis and Production Research program to MHM. Carlsberg Foundation is acknowledged for an instrument grant to acquire the ITC instrument. None of the funders took part in any aspect of the scientific content of the study.
Author information
Authors and Affiliations
Contributions
MAH and MHM conceived the study and designed the experiments. MHM performed all bioinformatics, molecular biology and biochemical characterization. LG along PB, RO and ET have contributed to the bio-electrochemical study. MAH and MHM wrote the first draft of the manuscript and all authors participated in writing and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Septoria cluster laccases. Figure S1. Chemical structure of the osmium polymer. Figure S2. Activity screening on aromatic (mainly phenolics) susbtrates. Figure S3. ZtrLac1A primary structure and purification. Figure S4. Cyclic voltammograms. Figure S5. Sequence alignment of ZtrLac1A and homologues from AA1_3. Figure S6. Architecture of Cu1 copper site in characterized ascomycetes laccases. (PDF 747 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Haddad Momeni, M., Bollella, P., Ortiz, R. et al. A novel starch-binding laccase from the wheat pathogen Zymoseptoria tritici highlights the functional diversity of ascomycete laccases. BMC Biotechnol 19, 61 (2019). https://doi.org/10.1186/s12896-019-0552-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12896-019-0552-4