
Abstract
Background
Mesenchymal chondrosarcoma (MCS) is a rare malignant variant of chondrosarcoma with a high tendency of recurrence and metastasis. Intradural extramedullary spinal MCS is exceedingly rare and usually found in pediatric patients. Herein, we present an elderly patient with primary intradural extramedullary spinal MCS. Relevant literatures are reviewed to disclose characteristics of intradural extramedullary spinal MCS.
Case presentation
A 64-year-old female presented with urinary difficulty and tightness of upper back preceding progressive weakness of right lower extremity. Magnetic resonance imaging revealed an intradural extramedullary tumor at the level of 3rd thoracic vertebra. This patient underwent total tumor resection and then received adjuvant radiotherapy. Histopathological examination showed that the tumor composed of spindle and round cells with high nucleocytoplasmic ratio accompanied by scattered eosinophilic chondroid matrix. Along with immunohistochemical findings and the existence of HEY1-NCOA2 fusion transcript, the diagnosis of MCS was confirmed. Neurologic deficit recovered nearly completely after surgery. No evidence of local recurrence or distant metastasis was found 5 years after treatments. Including the current case, a total of 18 cases have been reported in the literature with only one case with local recurrence and one case of mortality. The current case was the eldest patient diagnosed with primary intraspinal MCS in the literature.
Conclusions
MCS rarely appears in the intradural space of the spine. In contrast to classic MCS, treatment outcome of primary intradural extramedullary spinal MCS is usually excellent as total tumor resection is commonly achievable. Adjuvant radiotherapy may reduce local recurrence and chemotherapy may be associated with fewer recurrences especially for unresectable tumors.
Similar content being viewed by others

Background
Mesenchymal chondrosarcoma (MCS) is a rare malignant variant of chondrosarcoma whose incidence accounts for 0.2–0.7% of all malignant bone tumors or 3–10% of chondrosarcoma [1]. Even though a majority of these tumors are believed to be originated from bone, there is a considerable percentage around 33–50% that they can be detected in the extra-skeletal sites. Extra-skeletal MCSs most often involve the brain and meninges, occasionally intraspinal region [2]. Among them, intradural extramedullary MCS is exceedingly rare, which only has been described in sparse case reports with variable clinical traits. Recently, a novel fusion gene, HEY1-NCOA2, has been identified and adopted to confirm the diagnosis of MCS [3]. On the other hand, the vast majority of intradural extramedullary spinal tumors of adults are benign, in which the most common histological types are nerve sheath tumors, including schwannomas and neurofibromas, meningiomas, or ependymomas of the filum terminale [4]. MCS is a rare pathology involving the intradural extramedullary space of the spine. In this report, we document an adult case with primary intradural extramedullary MCS in the thoracic spine treated with surgical resection and adjuvant radiotherapy. Preoperative neurologic deficit recovered nearly completely after surgery. No local recurrence or distant metastasis was found 5 years after surgery.
Case presentation
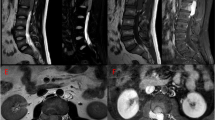
A 64-year-old woman initially experienced urinary difficulty and felt tightness at upper back. Weakness of right lower extremity developed 2 months later and rapidly progressed to inability of ambulation in following 2 weeks. She visited authors’ institute with presentation of monoplegia of right lower extremity and asymmetrically decreased response to light touch and pain below nipples, more prominent on the left side. Hyper-reflexia and positive Babinski sign at bilateral lower extremities were also noted. The results of routine blood tests were unremarkable and the patient had no family history of cancer or genetic disease. As the clinical manifestation was compatible with Brown-Séquard syndrome, a compressive lesion to spinal cord at the thoracic spine was suspected. Magnetic resonance imaging (MRI) revealed an intradural extramedullary mass at the level of 3rd thoracic (T3) vertebra with severe compression to the spinal cord (Fig. 1). The tumor was about 1.5 cm in size and characterized by intermediate signal intensity at T1-weighted images with mildly increased signal intensity at T2-weighted images and evident enhancement after the gadolinium administration. There was no bony involvement or other lesion found in radiologic assessments.

Preoperative MR images. a Sagittal T1-weighted image (T1WI), b Sagittal T2-weighted image (T2WI), c Sagittal T1WI with gadolinium enhancement, and d Axial T1WI with gadolinium enhancement. An intradural extrameduallary mass located at T3 level (arrows) and was characterized by intermediate signal intensity at T1WI, mildly increased signal intensity at T2WI and evident enhancement after gadolinium administration. Severe spinal cord compression by the tumor was shown (d)
The patient received surgical treatment for removal of the tumor mass on the next day after MRI study. During the surgery, the posterior surface of dural sac was exposed after total laminectomy from T2 to T4 vertebrae and then the intradural space was accessed through longitudinal opening of the dura mater. Grossly, the tumor sized 2 × 1.5 × 1.5 cm and was firm, reddish, lobulated, and hypervascular. The mass was attached to the inner surface of dura mater with no involvement of arachnoid or spinal cord. The tumor was removed en bloc after detached from dura mater. Posterior instrumentation and fusion were performed to prevent post-laminectomy kyphosis.
Histological examination revealed that the tumor was hypercellular and composed of spindle and round cells with a high nucleocytoplasmic ratio accompanied by scattered eosinophilic chondroid matrix (Fig. 2a). The tumor cells had ovoid to round nuclei and inconspicuous cytoplasm, arranged in vague fascicles (Fig. 2b). As for immunohistochemistry, the tumor cells were positive for CD99, desmin (especially on the chondrocyte-like cells) and CDK4 (focally), while negative for S100 protein, CK (AE1/AE3), CD34, MDM2, and myogenin. In addition, reverse transcription polymerase chain reaction (RT-PCR) was positive for HEY1-NCOA2 fusion transcript (Fig. 2c). Collectively, a mesenchymal chondrosarcoma of possible meningeal origin was diagnosed.

a At the × 100 magnification, the tumor is hypercellular and composed of spindle and round cells with a high nucleocytoplasmic ratio accompanied by scattered eosinophilic chondroid matrix. b At the × 200 magnification, the tumor cells have ovoid to round nuclei and inconspicuous cytoplasm, arranged in vague fascicles. Note the eosinophilic chondroid matrix (left field). c RT-PCR confirmed the presence of HEY1-NCOA2 fusion
The postoperative course was rather smooth with muscle power of right lower limb improved rapidly and completely after surgery. The patient received adjuvant radiation therapy with 44 Gray in 22 fractions. Follow-up MRI showed no evidence of recurrence 5 years after surgery and the patient remains now in complete remission, fully self-dependent with only mild sensory deficits at lower extremities (Additional file 1).
Discussion and conclusions
First described by Lichtenstein and Bernestein in 1959, MCS is a malignant tumor arising from bone or soft tissues [5]. Dowling reported the first case of MCS in non-osseous tissues in 1964 [6]. To date, an increasing number of reports suggest that MCS can occur anywhere in the body and at any age, with a male-to-female ratio of 1:1 and approximately 70% of the cases occurring during the second and the third decades of life [7].
Sparse cases of intradural MCS had been reported but there is not yet a systemic review in the literature. Compared to MCS at other anatomic sites, intradural MCS possesses a rather different clinical feature. Including the current case, the clinical information for 18 patients with primary intradural MCS in the literature is summarized in Table 1 [2, 8,9,10,11,12,13,14,15,16,17,18,19,20,21]. The current case is the eldest patient diagnosed with primary intraspinal MCS. Of all reported cases with primary intradural MCS, two-thirds of these patients were younger than 20 years old at the time of diagnosis. The male-to-female ratio was 1:1. The most prevalent symptoms were back pain and radicular pain, followed by sensory deficits, and muscle weakness. It was rare that urinary difficulty and Brown-Séquard syndrome were found as the initial manifestation. 50% (9/18) of intradural MCSs located in the thoracic spine, 17% (3/18) in the lumbar spine, 11% (2/18) in the cervical spine, 11% (2/18) in the thoracolumbar junction, and 11% (2/18) with disseminated lesions. As for the treatment, two-thirds of patients had gross tumor resection (12/18) and half of patients had adjuvant therapies (9/18). Only 1 case of local recurrence was reported during follow-up period (Case #16) and there was only 1 case of mortality (Case #10).
Histologically, most MCSs exhibit a biphasic pattern of islands of cartilage and areas of neoplastic small blue round cell component [5]. The exact histogenesis of intradural chondrosarcomas is still arguable. A probable hypothesis states that chondrosarcomas originate from primitive multipotential mesenchymal cells [22]. From the perspective of molecular pathology, the fusion gene encoding for the transcript HEY1-NCOA2 had been discovered in 2012 and have become a powerful tool for diagnosis. It showed both high sensitivity and high specificity since HEY1-NCOA2 was detected in nearly all cases of MCSs but not in other types of chondrosarcoma or Ewing sarcoma [3].
MRI remains the preferred imaging modality for intraspinal tumors, but there is no pathognomonic description for extra-osseous MCS. However, extra-osseous MCS typically present isointense signals with respect to the normal spinal cord on T1- weighted images while T2-weighted images show a high intensity or isointensity [23]. Besides, meningioma typically presents on MRI as a well-defined mass that is isointense to gray matter on T1-weighted images demonstrating avid enhancement after gadolinium administration, as in our case, making the differential diagnosis even more challenging [21]. Calcification can be seen occasionally but is believed to be not significantly related to the histologic findings and prognosis [23].
Radical surgery with complete removal of the tumor is considered the best choice of therapy for intradural MCS [13]. Clear resection margin predicts fewer local recurrences [24]. Due to the rarity of MCS, especially located in the intradural space, there is no general agreement on the necessity of adjuvant radiotherapy or chemotherapy. However, it has been demonstrated that adjuvant radiotherapy may reduce local recurrence [25] and chemotherapy may be associated with fewer recurrences especially for localized tumors [24]. From the aspect of intradural extramedullary tumors, even if total tumor resection is achieved during surgery, radiotherapy is indicated for intradural malignant tumors and chemotherapy is reserved for recurrent tumors with no other options in adult patients [4]. The dosage of post-operative radiation therapy was 44–78 Gray in previous studies [25]. The current case had adjuvant radiation therapy with 44 Gray in 22 fractions after the surgery of total tumor resection.
In general, the prognosis of MCS is poor regardless of the primary site of occurrence and 10-year survival rates in the literatures varying from 21 to 67% [26]. Local recurrence or distant metastases of MCS, particularly to lungs, lymph nodes and other bones, may appear even many years after the initial treatment [12]. Therefore, long-term follow-up is mandatory. In contrast to classic MCS, the prognosis of intradural extramedullary spinal MCS is remarkably better with only 1 case of local recurrence and 1 case of mortality among 18 cases reported in the literature. The superior treatment results of intradural extramedullary MCS maybe because complete resection of intradural extramedullary tumors is usually achievable and the neurologic prognosis after total tumor resection is often better than the other spinal neoplasia [4]. Some authors suggested that intraspinal MCS with dural attachment appeared to have a more favorable prognosis in comparison with those at other locations. It would be a result of early diagnosis and surgical intervention since neurologic deficit due to spinal cord compression would have already been noticed when the tumor is still small [7, 12].
In conclusion, MCS rarely located inside the dura sac of the spine. It occurs more frequently in pediatric patients and less in adults. In contrast to classic MCS, the functional recovery of intradural extramedullary spinal MCS is usually excellent as total tumor resection is commonly achievable. Adjuvant radiotherapy may reduce local recurrence and chemotherapy may be associated with fewer recurrences especially for unresectable tumors.
Availability of data and materials
Data sharing is not applicable to this case report as no datasets were generated or analyzed during the current study.
Abbreviations
- MCS:
-
Mesenchymal chondrosarcoma
References
Fletcher DM, Unni KK, Mertens F. World Health Organization classification of tumours. Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002.
Derenda M, Borof D, Kowalina I, Wesolowski W, Kloc W, Iżycka-Świeszewska E. Primary spinal intradural mesenchymal chondrosarcoma with several local regrowths treated with osteoplastic laminotomies: a case report. Surg J (NY). 2017;3:e117–23.
Wang L, Motoi T, Khanin R, Olshen A, Mertens F, Bridge J, et al. Identification of a novel recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosom Cancer. 2012;51:127–39.
Ogden AT, McCormick PC. Intradural extramedullary spinal tumors. In: Bridwell KH, DeWald RL, editors. The textbook of spinal surgery. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2011. p. 1535–43.
Lightenstein L, Bernstein D. Unusual benign and malignant chondroid tumors of bone. A survey of some mesenchymal cartilage tumors and malignant chondroblastic tumors, including a few multicentric ones, as well as many atypical benign chondroblastomas and chondromyxoid fibromas. Cancer. 1959;12:1142–57.
Dowling EA. Mesenchymal chondrosarcoma. J Bone Joint Surg Am. 1964;46:747–54.
Zibis AH, Wade Shrader M, Segal LS. Case report: mesenchymal chondrosarcoma of the lumbar spine in a child. Clin Orthop Relat Res. 2010;468:2288–94.
Scheithauer BW, Rubinstein LJ. Meningeal mesenchymal chondrosarcoma: report of 8 cases with review of the literature. Cancer. 1978;42:2744–52.
Lee ST, Lui TN, Tsai MD. Primary intraspinal dura mesenchymal chondrosarcoma. Surg Neurol. 1989;31:54–7.
Huckabee RE. Meningeal mesenchymal chondrosarcoma of the spine: a case report. J Magn Reson Imaging. 1991;1:93–5.
Ranjan A, Chacko G, Joseph T, Chandi SM. Intraspinal mesenchymal chondrosarcoma - case report. J Neurosurg. 1994;80:928–30.
Rushing EJ, Armonda RA, Ansari Q, Mena H. Mesenchymal chondrosarcoma: a clinicopathologic and flow cytometric study of 13 cases presenting in the central nervous system. Cancer. 1996;77:1884–91.
Li YH, Yao XH. Primary intradural mesenchymal chondrosarcoma of the spine in a child. Pediatr Radiol. 2007;37:1155–8.
Belhachmi A, Akhaddar A, Gazzaz M, Elasri C, Elmostarchid B, Boucetta M, et al. Primary spinal intradural mesenchymal chondrosarcoma. A pediatric case report. J Neuroradiol. 2008;35:189–91.
Sharma P, Ranjan A, Swarnalata Gowrishankar S, Lath R. Disseminated cranio-spinal intradural mesenchymal chondrosarcoma. Neurol India. 2012;60:252–4.
Turel MK, Rajshekhar V. Primary spinal extra-osseous intradural mesenchymal chondrosarcoma in a young boy. J Pediatr Neurosci. 2013;8:111–2.
Lee E, Lee HY, Choe G, Kim KJ, Lee WW, Kim SE. Extraskeletal intraspinal mesenchymal chondrosarcoma; 18F-FDG PET/CT finding. Clin Nucl Med. 2014;39:e64–6.
Andersson C, Osterlundh G, Enlund F, Kindblom LG, Hansson M. Primary spinal intradural mesenchymal chondrosarcoma with detection of fusion gene HEY1-NCOA2: a paediatric case report and review of the literature. Oncol Lett. 2014;8:1608–12.
Yang C, Fang J, Xu Y. Spinal extraosseous intradural mesenchymal chondrosarcoma. Spine J. 2016;16:e711.
Di Giannatale A, Colletti M, Russo I, Ferruzzi V, Dell’Anna VA, Cozza R, et al. Intraspinal mesenchymal chondrosarcoma: report of a pediatric case and literature review. Tumori. 2017;103(Suppl 1):S66–72.
Presutto E, Patel S, Fullmer J, Ezhapilli S. Extraosseous intradural chondrosarcoma of the cervical spine: a case report with brief review of literature. Case Rep Radiol. 2018;2018:6921020.
Vanderhooft JE, Conrad EU, Anderson PA, Richardson ML, Bruckner J. Intradural recurrence with chondrosarcoma of the spine. A case report and review of the literature. Clin Orthop Relat Res. 1993;294:90–5.
Bae GS, Choi SW, Youm JY, Kim SH. Primary spinal dumbbell-shaped mesenchymal chondrosarcoma located intradurally and extradurally. J Korean Neurosurg Soc. 2011;50:468–71.
Frezza AM, Cesari M, Baumhoer D, Biau D, Bielack S, Campanacci DA, et al. Mesenchymal chondrosarcoma: prognostic factors and outcome in 113 patients. A European musculoskeletal oncology society study. Eur J Cancer. 2015;51:374–81.
De Amorim BK, Liebsch N, Chen YL, Niemierko A, Schwab JH, Raskin K, et al. Clinical outcomes for patients after surgery and radiation therapy for mesenchymal chondrosarcomas. J Surg Oncol. 2016;114:982–6.
Dantonello TM, Int-Veen C, Leuschner I, Schuck A, Furtwaengler R, Claviez A, et al. Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: experiences of the CWS and COSS study groups. Cancer. 2008;112:2424–31.
Acknowledgements
Not applicable
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
Study concept and design: S-HY. Surgery performed by M-HH and S-HY. Participation in the initial management and follow-up: R-LH and S-HY. Analysis and interpretation of data: J-CL and H-YH. Review of the literature: I-HC. Drafting of the manuscript: C-WC and I-HC. Critical revision of the manuscript for important intellectual content: C-WC, M-HH, R-LH, and S-HY. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This case report was approved by the Institutional Review Board of National Taiwan University Hospital, Taipei, Taiwan. Patient’s informed consent was obtained. This case report has been conducted according to the principles expressed in the Declaration of Helsinki.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report, including any accompanying images. A copy of the consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Clinical timeline of our patient. (PDF 27 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chen, CW., Chen, IH., Hu, MH. et al. Primary intradural extramedullary spinal mesenchymal chondrosarcoma: case report and literature review. BMC Musculoskelet Disord 20, 408 (2019). https://doi.org/10.1186/s12891-019-2799-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12891-019-2799-2