
Abstract
Background
For the treatment of calcific tendinitis of the shoulder a variety of treatment regimes exist. Commonly used treatment measures include medication with oral analgesics, corticosteroid injections, extracorporeal shockwave therapy, ultrasound guided needling and lavage, and surgical treatment. Earlier cohort studies suggest that patients may benefit from these treatments, but there are few randomized studies and conflicting evidence about the effectiveness of the various treatments. In the present study we aim to compare the effectiveness of ultrasound guided needling and lavage (barbotage) together with a steroid injection to sham barbotage with and without an additional steroid injection.
Methods
The study will be performed in six secondary-care institutions in Norway and Sweden. It is designed as a pragmatic, randomized, three-arm, parallel group, double-blinded, sham-controlled clinical trial with a 2-year follow-up. It will be performed on 210 patients, aged 30 years or older, presenting with painful arc, positive impingement sign and a calcium deposit > 5 mm. Randomization to one of the three treatment options will be performed by using an online central randomization system. The three treatment groups are barbotage together with a subacromial steroid injection (the barbotage group), sham barbotage together with a subacromial steroid injection (the steroid group) or sham barbotage without a subacromial steroid injection (the placebo group). In the placebo group the steroid injection will be replaced by a short-acting local anaesthetic. Standardized home-based post-treatment physiotherapy will be performed by all patients for 8 weeks. Follow-ups are at 2 and 6 weeks, 4, 8, 12 and 24 months after treatment was given and will be performed with the patients and the outcome assessors blinded for group assignment. Primary outcome will be the Oxford shoulder score at 4 month follow-up. Secondary outcome measures are the QuickDASH upper extremity score, the EQ-5D-5L general health score and visual analogue scales for pain at rest, during activity, and at night.
Discussion
The scientific evidence from this placebo-controlled trial will be of importance for future treatment recommendations in patients with calcific tendinitis.
Trial registration
ClinicalTrials.gov: NCT02419040, registered 10 April 2015
EudraCT: 2015-002343-34, registered 23 September 2015 (retrospectively registered)
Similar content being viewed by others


Background
Calcific tendinitis is a painful disorder of the shoulder of unknown etiology [1]. The condition is characterized by the formation of deposits of calcium crystals in one or several of the rotator cuff tendons. The prevalence of the condition has been reported to be 3 to 10% in the general population [2, 3] and 7 to 17% in individuals with shoulder pain [4]. Tendon inflammation located around the deposit is considered to contribute to pain. The course of the disease is often self-limiting with spontaneous calcium resorption and resolution of symptoms over several months [3]. In cases in which resorption is delayed, absent or incomplete, symptoms may persist and anti-inflammatory treatment and/or removal of the calcification may provide symptomatic relief. Different treatment methods including anti-inflammatory and analgesic medication, extracorporal shockwave therapy (ESWT), ultrasound guided needling and lavage (barbotage), and surgical treatment are in use, but a consensus on the preferred treatment is lacking.
Farin introduced barbotage as an ultrasound guided technique in 1995 [5]. It consists of needle aspiration and lavage of the calcium deposit. Both single and double needle procedures have been described [5–9]. Good short- and medium-term results have been reported from several cohort studies [6, 7, 10] . A systematic review of the efficacy of barbotage in the treatment of calcific tendinitis found the technique to be safe and effective with an estimated average of pain improvement of 55% [11]. Only few comparison studies between barbotage and other techniques have been performed. In one randomized study, barbotage was found to be superior to subacromial corticosteroid treatment [8]. Contrary, a systematic review and meta-analysis of minimally invasive therapies in the management of chronic calcific tendinopathy, concluded that barbotage was not more effective than subacromial corticosteroid injections. The review concluded that further research is needed to evaluate its effectiveness [12]. Considering the cyclic often self-limiting course of the disease, and the treatment’s anticipated placebo effect, the effectiveness of barbotage should preferably be answered by a sham-controlled randomized study.
Aims
The aim of the present study is:
-
(1)
to compare the short-term effectiveness (at 4 months) of barbotage and steroid injection, sham barbotage and steroid injection, and sham barbotage without steroid injection in the treatment of calcific tendinitis, and
-
(2)
to compare the long-term effectiveness (2 years) between the study groups.
Methods/Design
Trial design
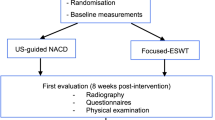
The study will be performed at five hospitals in Norway and one hospital in Sweden as a pragmatic, randomized, three-arm, parallel group, double-blinded, sham-controlled superiority trial with a 2-year follow-up (Fig. 1). Departments of orthopaedics, radiology and physical medicine and rehabilitation will be involved in the conduction of the study. The recruiting sites are Martina Hansens Hospital, Sandvika; Helse Fonna Hospital, Stord; Haraldsplass Deaconess Hospital, Bergen; Vestfold Hospital, Stavern; Oslo University Hospital, Oslo; (all Norway), and Linköping University Hospital, Linköping, (Sweden). The population group that will be considered for study participation consists of patients referred from general practitioners from the catchment areas of the study hospitals. They represent the unselected group of patients with painful calcific tendinitis with inadequate effect from treatment measures performed in general practice care. Patients who will have to be excluded according to the criteria given in Tables 1 and 2 will be reported. Inclusion, treatment and follow-up of study patients will be performed by hospital based physicians, specialised in physical medicine or orthopaedic surgery, all with long experience and special interest in the treatment of shoulder patients. At one site the study intervention will be given by a radiologist. The study hospitals represent different types of hospitals ranging from university hospitals to municipal hospitals and from hospitals with an urban to those with a rural location. Study medication and equipment is available at all study sites as all study interventions are part of already existing treatment routines. The study will be conducted in compliance with the principles of the Declaration of Helsinki, the principles of Good Clinical Practice (GCP) and under consideration of national laws and regulations, and will be reported in accordance with the CONSORT guidelines.

Patient flow through the study
Participants
Study participants will be recruited at the trial centers among patients referred from primary care services for treatment of a painful shoulder and in whom calcific tendinitis was diagnosed after clinical and radiological assessment. Eligibility for inclusion will be based on the presence of the inclusion criteria and the absence of the exclusion criteria given in Tables 1 and 2.
Data collection at baseline
The following demographic and baseline data will be obtained at the primary consultation: Gender, age, shoulder affected (right, left, both), hand dominance, duration of symptoms, earlier treatment, use of analgesics, use of concomitant medication, comorbidities, occupational status, shoulder demanding activities at work or leisure time, and smoking habits. Standard shoulder radiographs (anterior-posterior, lateral and acromioclavicular views) will be obtained not more than 4 weeks prior to the intervention. The size and the type of the deposit will be recorded according to the Molé classification [13]. Diagnostic ultrasound of both shoulders will be performed and the size and the location of the deposits, related to the long head of the biceps tendon, will be recorded. On the treatment day, prior to treatment, three patient related outcome scores (Oxford Shoulder Score (OSS) [14, 15], quickDASH upper extremity score [16, 17], EQ-5D-5L general health score [18, 19]) and three visual analogue scales (VAS) for pain at rest, during activity and at night will be completed by the patient, together with the Stanford expectations of treatment scale (SETS) [20].
All study scores exist in a digital version in both languages. Patients fill in the scores directly on a computer/tablet through an internet website which is encrypted and has a secure protocol. Baseline and follow-up data will be stored on the research server at Martina Hansens Hospital, Sandvika, Norway. Access to the server is password protected and only the principal investigator (SM) has access to the database.
Randomisation
Patients who fulfil the inclusion and exclusion criteria and who have signed the consent form after oral and written study information was given, will be randomized to one of the three treatment options. Randomization will take place on the day of the intervention by an online central randomization system (web-CRF) developed and administered by the Unit of Applied Cancer Research, Institute of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway. To assure verifiability of the randomization, patient initials, year of birth and name of the treating hospital will have to be registered at the central randomisation database. After registration, the intervention group for the specific patient will be displayed on-screen. Allocation will be 1:1:1. Block randomization with varying block lengths and stratification according to hospital will be performed. The randomisation list will remain at the University of Trondheim for the whole duration of the study and, consequently, will be inaccessible for the investigators, care providers and outcome assessors at the study centres.
Intervention
An orthopaedic surgeon (2 hospitals), a radiologist (1 hospital), or a specialist in physical medicine (3 hospitals), all with at least 5 years of experience in diagnostic and interventional ultrasound of the shoulder will conduct the allocated interventions. To ensure consistency a video of the procedure was produced and send to all trial sites (Additional file 1: Video barbotage procedure).
Interventions will be performed with the patient in supine position. The arm will be internally rotated to a degree that the treating physician considers to be the most favourable for the puncturing of the calcific deposit (usually arm on the back). An opaque sheet will be used to block the patient’s view of the screen of the ultrasound machine. After sterile skin preparation, with a 7 to 14 MHz transducer wrapped in a sterile drape, and by using sterile jelly, the calcific deposit will be sonographically identified, usually on the lateral transversal view. A 21-gauge needle will be introduced into the shoulder under sonographic guidance, and the pathway and the subacromial-subdeltoid bursa will be anesthetized with an injection of 10 ml of 1% lidocaine hydrochloride with adrenaline 5 μg/ml.
In patients randomized to barbotage a new 18-gauge needle connected to a 5 ml syringe with 4 ml of saline solution will be used to puncture the calcification using freehand technique under continuous sonographic monitoring. With the tip of the needle placed in the centre of the deposit, the calcification will be flushed. If backflow of calcific material can be identified in the syringe, lavage of the deposit will be performed by successive propulsion and aspiration with the syringe plunger. Extracted calcium can be visualized in the syringe as a cloudlike substance that settles at the bottom of the barrel. To avoid reinjection of the calcium into the deposit, the needle and the syringe will be hold in the horizontal plane during the procedure. The syringe will be substituted when the fluid has become cloudy and the procedure will be repeated until the backflow becomes clear. At the end of the procedure the content of the syringes will be poured into a measuring cup and, when settled at the bottom of the cup, the volume of the extracted calcium will be estimated. In cases where no material can be extracted, repeated perforation of the deposit will be performed to possibly initiate or accelerate spontaneous resorption.
In patients randomized to the steroid group or to the placebo group, the tip of the 18-gauge needle will be placed in the soft parts outside of the rotator cuff and movements mimicking the lavage procedure will be performed. A lavage procedure usually takes 5 min and a similar time period will be used for the mimicking manoeuvre.
Finally in all three groups, under sonographic monitoring, a new 21-gauge needle will be introduced into the subacromial bursa and 9 ml of Lidocaine hydrochloride 10 mg/ml and 1 ml of Triamcinolone 20 mg/ml (in the barbotage and the steroid group) or 10 ml of Lidocaine hydrochloride 10 mg/ml (in the placebo group) will be injected into the subacromial bursa.
Blinding
The study will be conducted double-blinded with masking of patients and outcome assessors for treatment selection. We will blind the patient during the procedure by blocking their view of the ultrasound screen and the doctors working space and by mimicking a barbotage procedure in all patients. To control whether blinding of the patients was successful, patients will be asked after the treatment, and again after 2 and 6 weeks, which treatment they believe was performed. A blinding index as given in the literature will be calculated [21].
The specialists performing the study interventions will be excluded from all other tasks during the follow-up and the statistician conducting the statistical analysis will be blinded to the treatment given.
The specific treatment procedure will not be reported in the patient journal, only that the patient received the study treatment, according to randomisation. The specialist who performs the study treatment, will record the patient’s randomisation number, treatment selection, and name and date of birth in a decryption list which is kept in a safe place, accessible to the treating doctor only. This list can be consulted only if code break should be necessary during the trial. Code break is only permitted if knowledge of the study treatment seems mandatory for further treatment of a patient or if the patient insists in knowing the treatment group. All code breaks together with their causes will be recorded.
Post-intervention treatment
In case of post interventional pain exacerbation non-prescription analgesics are recommended.
Routine use of the shoulder will be allowed without restrictions, but the patient is asked to avoid heavy shoulder labour for 2 weeks.
About 1 week after the intervention all patients will start on a standardized home-based physiotherapeutic treatment regime. The program consists of four shoulder exercises, and will be presented by written instructions, on photographic illustrations (Additional file 2: Folder physiotherapy procedure), and as a video (http://youtu.be/6nRYdqYniUI) [22]. Prior to the start of the program, a physiotherapist at each hospital will teach the patients how to perform the exercises correctly. All patients will have to maintain a regular protocol over 8 weeks during which they record each training session with date and number of exercises performed.
Data collection at follow-up
Follow-up data will be collected 2 and 6 weeks, 4, 8, 12 and 24 months after the intervention (Table 1). At each time point the OSS [14, 15], the QuickDASH upper extremity score [16, 17], the EQ-5D-5L general health score [18, 19] and VAS for pain at rest, during activity, and at night will be filled in. To perform the 2 and 6 week and 8 and 12 month follow-ups patients will receive an e-mail with a link to a website, where they can fill in the study questionnaires at their computer at home.
At clinical follow-ups after 4 and 24 months study scores will be filled in on a computer/tablet at the hospital. The blinded assessor will register all treatment related adverse events by asking and by consulting each patient’s adverse event diary. The adverse event diary will be handed out to the patients on treatment day and will have to be kept over the entire 24-month follow-up period. Patients will be asked to enter in the diary any changes of their health condition that theiy perceive as an adverse event. The type of the event, its date of occurrence, its duration and its severity (on a 5 point Likert scale ranging from mild to severe) will have to be given. In case of a serious event, the hospital will have to be contacted immediately. If an adverse event requires treatment this will be recorded by the physician who is responsible for patient follow-up. The use and the dosage of prescription analgesics during the post treatment period will be recorded. An X-ray of the shoulder will be performed and, if still visible, the calcific deposit will be measured and classified according to Molé [13], blinded for baseline results.
Outcome assessment
The study’s primary outcome measure is the OSS with the outcome at 4 months as the result of primary interest. Secondary outcomes are the results on the OSS at the other points of follow-up, results of the other study scores at all points of follow-up and the number of patients in each treatment group who change treatment during the study.
Scoring instruments
All scoring instruments which will be used in the study are patient related outcome measures.
The OSS [14, 15] is a validated shoulder specific, 12-item score. It contains questions about pain and function and has been shown to be responsive to both surgical and non-surgical interventions on the shoulder. Its scoring method was modified in 2009 so that each of the 12 questions has five response categories scored from 4 (best) to 0 (worst) resulting in a total score ranging from 0 to 48 with a lower result indicating a greater degree of disability [15].
The QuickDASH upper extremity score [16, 17] is a shortened version of the DASH score with the number of items reduced from 30 to 11. It uses 5-point Likert scales to assess physical function and symptoms. It includes two domains for sport/art and work that are scored separately. The obtained value is transformed to a score ranging from 0 to 100 with a higher score indicating greater disability.
The Norwegian translation of the OSS and the Norwegian and Swedish version of the DASH score have been validated [23–25].
The EQ-5D-5L [18, 19] measures general health related quality of life. It comprises the dimensions mobility, self-care, usual activities, pain/discomfort, anxiety/depression each with 5 response categories, ranging from no problems to extreme problems. The score further includes a visual analogue scale for assessment of the health condition, ranging from the worst health (0) to the best health (100) you can imagine. Results can be presented as a health profile or as an index value.
Shoulder pain over the last week will be measured on three 0–10 visual analogue scales for pain at rest, during activities and at night. The scales are labelled no pain at the left end and worst imaginable pain at the right end.
The SETS [20] is a quick and easy-to-administer tool for the measurement of positive and negative pretreatment expectancies. The score consists of six items, three of them measuring positive expectancy, and three of them negative expectancy. It can be used to assess the influence of pretreatment expectancies on the outcomes in trials comparing real and sham treatment.
Change of treatment
Patients who are still symptomatic or have redeveloped symptoms at four month follow-up or later, will be considered for supplementary treatment measures by the blinded follow-up assessor. If necessary, they will be offered treatment as usual, which means barbotage (repeated barbotage if barbotage was the primary treatment), steroid injection (repeated injection if injection was the primary treatment), ESWT, surgery, or therapist guided physiotherapy treatment, depending on findings and patient preferences. Earlier re-examination and change of treatment can be considered, in accordance with the Helsinki Declaration, for patients who are symptomatic to an extent that they cannot wait until the four month control. Patients who change the treatment during follow-up will remain in the study and will be followed-up according to an intention-to-treat principle. Unblinding of patients in conjunction with a change of treatment, will only be performed if the patient explicitly insists on knowing his primary treatment selection.
Concomitant pain management
Previous treatment with analgesics is allowed but has to be stopped 48 h prior to baseline. In the post-treatment period the use of non-prescription analgesics such as Paracetamol (500 mg), Ibuprofen (600 mg) or Naproxen (250 mg) is permitted as it is considered part of the treatment. If post-treatment pain management necessitates the use of prescription analgesics the type and dosage will be recorded in the patients case report form at 4 and 24 month follow-ups.
Harms
An adverse event diary will include all occurrences that the patient perceives as an adverse event. At follow-up, adverse events will be recorded by the follow-up assessor and will be sent to the sponsor. Once a year throughout the clinical trial, the sponsor will provide the respective Medicines Agency with an annual safety report. The format will comply with national requirements. If serious adverse events should occur, the sponsor will be informed immediately by phone or email.
Sample size
Calculation of sample size was performed for an ANOVA of our primary outcome, which is the result on the OSS at 4 month follow-up. To detect a minimally important difference of 4 (SD 7) points [26] with a power of 90%, a 2-sided significance level of 0.05, 60 patients are required in each treatment group. To compensate for expected 15% drop-outs, we plan to include 70 patients in each treatment group. A supplementary analysis showed that this sample size also will be sufficient for pairwise post hoc t-test analyses (1 versus 2, 2 versus 3 and 1 versus 3), still with a significance level after Bonferroni correction of 0.017 but with a power of approximately 80%.
The actual statistical analysis of the interventions on primary outcome will be conducted using linear mixed models for repeated measurements adjusted for outcome measure at baseline. It is expected to give a slightly higher statistical power than an ANOVA.
Statistical analysis
Demographic baseline data will be expressed for categorical variables as number of cases and for continuous variables by means with SD (if normally distributed) or by medians with range (if not normally distributed).
A linear mixed model for repeated measurements will be used for analysis. Because of an expected large number of crossovers after 4 months, the primary analysis will be performed on results up to 4 month follow-up and a secondary analysis on results up to 24 month follow-up. Analyses will be performed adjusted for baseline differences of the OSS and according to intention-to-treat. The linear mixed model will be estimated using linear maximum likelihood and include a random intercept, measure of the OSS at baseline as a covariate and observation time after intervention and type of intervention as factors. Mean differences (95% CI) between groups at 4 months follow-up will be presented from the linear mixed model to assess difference between interventions. A 2-sided p-value ≤ 0.05 is considered significant. Post-hoc pairwise comparisons for the primary outcome will be performed with p-value adjustments according to Bonferroni. Missing values will be handled by using mixed model analysis. Supplementary per-protocol analyses will be performed. A similar statistical analysis as described for the OSS will be used for the continuous secondary outcomes (QuickDASH, EQ-5D-5L).
Categorical variables will be expressed as numbers and percentages, and differences between groups will be analysed by the Chi2 or Fisher’s exact test. Possible associations between categorical baseline variables and outcomes will be explored by logistic regression analysis.
All subgroup analyses will be exploratory in nature and are planned for the size of the calcification at baseline (≤12.5 mm versus >12.5 mm), the volume of the removed material (≤0.1 ml versus > 0.1 ml), and the results on the Stanford expectation of treatment scale (positive expectancies versus negative expectancies). Selection of the threshold values for the size of the calcification at baseline and for the volume of the removed material are based on the findings of an unpublished pilot study from our institution.
Clinical safety will be investigated by assessing adverse events in a descriptive manner.
The statistician responsible for data analyses will blinded from the treatment allocation until completion of analyses.
Discussion
Treatment of patients with long-standing symptoms from calcific tendinitis is controversial. If primary non-invasive treatment fails, different mini-invasive or non-invasive treatment options such as injection therapy, barbotage or ESWT exist. Which of them should be preferred is unclear.
Sham studies aiming to assess the effectiveness of invasive interventions for shoulder disorders are rarely performed but are necessary to provide a better understanding of the therapeutic mechanisms. In the present study we want to compare two mini-invasive approaches representing different therapeutic principles; in the steroid injection group symptomatic relieve will be tried to achieve by anti-inflammatory treatment alone and in the barbotage group by a supplementary removal of the calcific deposit. A literature search identified only one randomised study comparing these methods [8]. The study was performed on 48 patients and showed a significantly better result on the Constant score for the barbotage group at one year follow-up (86.0 versus 73.9 points). The present study will be based on a larger patient group and a longer follow-up, and will also include a placebo group. Inclusion of a placebo group is important as we aim to assess the contribution of the placebo response to treatment results. Comparison to placebo has traditionally been performed in drug studies. However, the existence of a surgical placebo effect can be assumed and may be underestimated [27]. This would apply in particular for the treatment of conditions where pain is the dominant symptom, as pain is the outcome most powerfully affected by placebo interventions [28]. Inclusion of a placebo group as a comparator in studies assessing the effectiveness of invasive or mini-invasive procedures has increased in recent years [29] and has revealed that the results of some surgical procedures are not different from placebo [30, 31]. Without a placebo group we cannot exclude that patients treated by cortisone injection or barbotage are exposed to an ineffective mini-invasive procedure with a profound placebo effect.
To ensure recruitment of a sufficient number of eligible patients, the study will be performed at six hospitals as a multicenter study. Based on the number of barbotage procedures performed at these hospitals in the time before study start, inclusion of ten patients per year and per site can be expected. To minimise the number of drop-outs, data capturing at four of the six follow-ups will be via email where the patients can fill in the study scores on a computer at home. Non-appearance of follow-up data will be noticed immediately on the central server and can be handled by sending out a reminder.
The aim of the present study is to contribute to better knowledge about the mechanisms for pain reduction and improvement of function in the treatment of patients with symptomatic calcific tendinitis.
Abbreviations
- ESWT:
-
Extracorporal shock wave therapy
- GCP:
-
Good Clinical Practice
- OSS:
-
Oxford shoulder score
- SD:
-
Standard Deviation
- SETS:
-
Stanford expectation of treatment score
- VAS:
-
Visual analogue scale
References
Jerosch J, Strauss JM, Schmiel S. Arthroscopic treatment of calcific tendinitis of the shoulder. J Shoulder Elbow Surg. 1998;7(1):30–7.
Bosworth BM. Calcium deposits in the shoulder and subacromial bursitis. A survey of 12122 shoulders. JAMA. 1941;116(22):2477–82.
Uhthoff HK, Loehr JW. Calcific tendinopathy of the rotator cuff: pathogenesis, diagnosis and management. J Am Acad Orthop Surg. 1997;5(4):183–91.
Hedtmann A, Fett H. Die sogenannte periarthropathia humeroscapularis - Klassifizierung und Analyse anhand von 1266 Fällen. Z Orthop. 1989;127(6):643–9.
Farin PU, Jaroma H, Soimakallio S. Rotator cuff calcifications: treatment with US-guided technique. Radiology. 1995;195(3):841–3.
del Cura JL, Torre I, Zabala R, Legórburu A. Sonographically guided percutaneous needle lavage in calcific tendinitis of the shoulder: Short- and Long-term results. AJR Am J Roentgenol. 2007;189(3):W128–34.
Aina R, Cardinal E, Bureau NJ, Aubin B, Brassard P. Calcific shoulder tendinitis: treatment with modified US-guided fine-needle technique. Radiology. 2001;221(2):455–61.
de Witte PB, Selten JW, Navas A, Nagels J, Visser CP, Nelissen RG, et al. Calcific tendinitis of the rotator cuff. A randomized controlled trial of ultrasoundguided needling and lavage versus subacromial corticosteroids. Am J Sports Med. 2013;41(7):1665–73.
Sconfienza LM, Viganò S, Martini C, Aliprandi A, Randelli P, Serafini G, et al. Double-needle ultrasound-guided percutaneous treatment of rotator cuff calcific tendinitis: tips & tricks. Skeletal Radiol. 2013;42(1):19–24.
Yoo JC, Koh KH, Park WH, Park JC, Kim SM, Yoon YC. The outcome of ultrasound-guided needle decompression and steroid injection in calcific tendinitis. J Shoulder Elbow Surg. 2010;19(4):596–600.
Lanza E, Banfi G, Serafini G, Lacelli F, Orlandi D, Bandirali M, et al. Ultrasound-guided percutaneous irrigation in rotator cuff calcific tendinopathy: what is the evidence? A systematic review with proposals for future reporting. Eur Radiol. 2015;25(7):2176–83.
Louwerens JK, Sierevelt IN, van Noort A, van den Bekerom MP. Evidence for minimally invasive therapies in the management of chronic calcific tendinopathy of the rotator cuff: a systematic review and meta-analysis. J Shoulder Elbow Surg. 2014;23(8):1240–9.
Molé D, Kempf JF, Gleyze P, Rio B, Bonnomet F, Walch G. Resultat du traitement arthroscopique des tendinopathies non rompues, Il: les calcifications. Rev Chir Orthop. 1993;79(3):532–41.
Dawson J, Fitzpatrick R, Carr A. Questionnaire on the perceprions of patients about shoulder surgery. J Bone Joint Surg Br. 1996;78(4):593–600.
Dawson J, Rogers K, Fitzpatrick R, Carr A. The Oxford shoulder score revisited. Arch Orthop Trauma Surg. 2009;129(1):119–23.
Hudak P, Amadio PC, Bombardier C, the Upper Extremity Collaborative Group. Development of an Upper Extremity Outcome Measure: the DASH (Disabilities of the Arm, Shoulder, and Hand). Am J Ind Med. 1996;29(6):602–8.
Beaton DE, Wright JG, Katz JN, the Upper Extremity Collaborative Group. Development of the QuickDASH: comparison of three item-reduction approaches. J Bone Joint Surg Am. 2005;87(5):1038–46.
EuroQolGroup. EuroQol: a new facility for the measurement of health related quality of life. Health Policy. 1990;16(3):199–208.
Herdman M, Gudex C, Lloyd A, Janssen MF, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res. 2011;20(10):1727–36.
Younger J, Gandhi V, Hubbard E, Mackey S. Development of the Stanford Expectations of Treatment Scale (SETS): a tool for measuring patient outcome expectancy in clinical trials. Clin Trials. 2012;9(6):767–76.
James KE, Bloch DA, Lee KK, Kraemer HC, Fuller RK. An index for assessing blindness in a multi-centre clinical trial: disulfiram for alcohol cessation - a va cooperative study. Stat Med. 1996;15(13):1421–34.
Moosmayer S, Seljom US, Gärtner AV. Home based exercises for the shoulder. 2015. http://youtu.be/6nRYdqYniUI. Accessed 7 Nov 2016.
Ekeberg OM, Bautz-Holter E, Tveitå EK, Keller A, Juel NG, Brox JI. Agreement, reliability and validity in 3 shoulder questionnaires in patients with rotator cuff disease. BMC Musculoskelet Disord. 2008;9:68. doi:10.1186/1471-2474-9-68.
Haldorsen B, Svege I, Roe Y, Bergland A. Reliability and validity of the Norwegian version of the Disabilities of the Arm, Shoulder and Hand questionnaire in patients with shoulder impingement syndrome. BMC Musculoskelet Disord. 2014;15:78. doi:10.1186/1471-2474-15-78.
Atroshi I, Gummesson C, Andersson B, Dahlgren E, Johansson A. The disabilities of the arm, shoulder and hand (DASH) outcome questionnaire: reliability and validity of the Swedish version evaluated in 176 patients. Acta Orthop Scand. 2000;71(6):613–8.
van Kampen DA, Willems JW, van Beers L, Castelein RM, Scholtes V, Terwee CB. Determination and comparison of the smallest detectable change (SDC) and the minimal important change (MIC) of four-shoulder patient-reported outcome measures (PROMs). J Orthop Surg Res. 2013;14(8):40. doi:10.1186/1749-799X-8-40.
Holtedahl R, Brox JI, Tjomsland O. Placebo effects in trials evaluating 12 selected minimally invasive interventions: a systematic review and mveta-analysis. BMJ Open. 2015;5(1):e007331. doi:10.1136/bmjopen-2014-007331.
Hróbjartsson A, Gøtzsche PC. Is the placebo powerless? An analysis of clinical trials comparing placebo with no treatment. N Engl J Med. 2001;344(21):1594–602.
Wartolowska K, Collins GS, Hopewell S, Judge A, Dean BJ, Rombach I, et al. Feasibility of surgical randomised controlled trials with a placebo arm: a systematic review. BMJ Open. 2016;6(3):e010194. doi:10.1136/bmjopen-2015-010194.
Moseley JB, O’Malley K, Petersen NJ, Menke TJ, Brody BA, Kuykendall DH, et al. A controlled trial of arthroscopic surgery for osteoarthritis of the knee. N Engl J Med. 2002;347(2):81–8.
Sihvonen R, Paavola M, Malmivaara A, Itälä A, Joukainen A, Nurmi H, Finnish Degenerative Meniscal Lesion Study (FIDELITY) Group, et al. Arthroscopic partial meniscectomy versus sham surgery for a degenerative meniscal tear. N Engl J Med. 2013;369(26):2515–24.
Kessel L, Watson M. The painful arc syndrome. Clinical classification as a guide to management. J Bone Joint Surg Br. 1977;59(2):166–72.
Hawkins RJ, Kennedy JC. Impingement syndrome in athletes. Am J Sports Med. 1980;8(3):151–8.
Neer 2nd CS. Impingement lesions. Clin Orthop Relat Res. 1983;173:70–7.
Acknowledgements
The authors would like to thank the surgical directors, colleagues and staff at all study hospitals, who allowed us to conduct this trial.
We would also like to thank the members of the KALK study group for their contributions in the planning and conduction of the study: Unni S. Seljom, PT, and Anne V. Gärtner, PT, (Martina Hansens Hospital, Norway) for the development of the study’s physiotherapeutic treatment regime and Nina J. Kise, M.D. (Martina Hansens Hospital, Norway), Line K. Løvereide, M.D. (Stavern, Norway), Dalia Martinkiene, M.D. (Helse Fonna, Norway) and Ida Dånmark, M.D. (Linköping University Hospital, Sweden) for blinded follow-up of study patients.
Funding
The study received grants from Bergesenstiftelsen, Aase Bye and Trygve J.B. Hoffs fond, and Smith & Nephew. The funding did not play a role in the design of the study or in the writing of this manuscript.
Availability of data and materials
Not applicable.
Authors’ contributions
SM is the principal investigator. SM and OME designed the study and drafted the protocol. JB, SHK, IH, SK, JIB, NGJ, HBH and AHP all have made substantial contributions to the conception, design and coordination of the study. AHP has performed the sample size calculation and planned the statistical analyses. All authors and the KALK study group were involved in drafting of the present manuscript and have revised the manuscript critically for important intellectual content and have read and approved the final version to be published.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
We confirm that written informed consent was obtained from all subjects and researchers whose images are provided in the supplementary materials of this protocol.
Ethics approval and consent to participate
Approvals from the ethical committee and from the medical products agency have been received in Norway (Regional Committees for Medical and Health Research ethics, Ref. 2014/1183; Norwegian Medicines Agency, Ref. 15/08624-6) and Sweden (Regional ethical review board Linköping, Ref. 2015/7931; Medical Products Agency, Sweden, Ref. 5.1-2015-93735). Written informed consent will be obtained from all participants prior to inclusion.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional files
Video barbotage procedure. (AVI 256234 kb)
Additional file 2:
Folder physiotherapy procedure. (DOCX 490 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Moosmayer, S., Ekeberg, O.M., Hallgren, H.B. et al. KALK study: ultrasound guided needling and lavage (barbotage) with steroid injection versus sham barbotage with and without steroid injection - protocol for a randomized, double-blinded, controlled, multicenter study. BMC Musculoskelet Disord 18, 138 (2017). https://doi.org/10.1186/s12891-017-1501-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12891-017-1501-9