
Abstract
Bronchiectasis is a common feature of severe inherited and acquired pulmonary disease conditions. Among inherited diseases, cystic fibrosis (CF) is the major disorder associated with bronchiectasis, while acquired conditions frequently featuring bronchiectasis include post-infective bronchiectasis and chronic obstructive pulmonary disease (COPD). Mechanistically, bronchiectasis is driven by a complex interplay of inflammation and infection with neutrophilic inflammation playing a predominant role. The clinical characterization and management of bronchiectasis should involve a precise diagnostic workup, tailored therapeutic strategies and pulmonary imaging that has become an essential tool for the diagnosis and follow-up of bronchiectasis. Prospective future studies are required to optimize the diagnostic and therapeutic management of bronchiectasis, particularly in heterogeneous non-CF bronchiectasis populations.
Similar content being viewed by others


Background
Bronchiectasis is a condition in which an area of the bronchial lumen is permanently and abnormally widened, with accompanying infection. Bronchiectasis is found in a variety of pulmonary diseases, both genetically caused and acquired, such as severe pulmonary infections and cystic fibrosis (CF), but is also a feature of Kartagener syndrome, chronic obstructive pulmonary diseases (COPD), alpha 1-antitrypsin deficiency, asthma, or primary immunodeficiencies [1,2,3]. Bronchietasis is caused by long-term excessive inflammatory damage to the airways, which results in tissue breakdown, enlargement of the affected airways and the key clinical symptoms of chronic productive cough and shortness of breath. Globally, in up to half of all cases the cause cannot be identified (idiopathic). Those cases together with several other known aetiologies such as post-infectious and allergic hypersensitivity collectively fall under the category ‘non-cystic fibrosis’ (non-CF) bronchiectasis [4]. Here we discuss the key features of both CF and non-CF related bronchiectasis with respect to their pathogenesis, imaging and clinical management.
Pathogenesis of bronchiectasis formation
Bronchiectasis mechanistically results from chronic inflammatory microenvironments that trigger airway tissue breakdown. In both CF and non-CF bronchiectasis, the complex interplay between infection and inflammation feeds a pro-inflammatory vicious circle that progressively drives the generation of bronchiectasis and the destruction of the pulmonary architecture [5]. Inflammatory immune cells (mainly activated macrophages and neutrophils) represent the major infiltrating population in disease conditions associated with bronchiectasis and contribute significantly to tissue damage and bronchiectasis generation through the release of their harmful cellular ingredients. Particularly, cell-derived proteases and reactive oxygen species represent key mediators in the degradation and destruction of extracellular pulmonary tissue components, leading to bronchiectasis formation. The precise early immune-mediated mechanisms that trigger and maintain the formation of bronchiectasis remain yet incompletely understood. Regulated immune homeostasis seems to be essential since both immune deficiencies as well as hyper-active immune responses are associated with bronchiectasis. Particularly, the protease-antiprotease imbalance [6, 7], as found in CF and COPD airways, is considered as key pathogenic component in degrading extracellular matrix. Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene are causative for CF lung disease and drive the earliest pathogenic events in epithelial cells that ultimately lead to the genesis of bronchiectasis. Also beyond CF lung disease, CFTR-related cellular mechanisms regulating mucociliary clearance have been involved in cigarette smoke-induced COPD [8].
In the following two sections, we will focus on the microbiological (a) and immunological/inflammatory (b) findings associated with the pathogenesis of bronchiectasis.
Microbiology
Pseudomonas aeruginosa is a common and dominant pathogen found in the airways of both CF and non-CF bronchiectasis patients [9,10,11,12,13]. Chronic infection has been associated with more severe decline in lung function [14,15,16,17,18,19], increased hospitalizations [20, 21], frequent exacerbations [22] and disease severity [23, 24]. Although clinical manifestations between the two settings vary, their core airway microbiota is largely analogous [25]. Along with Pseudomonas, bacteria belonging to other genera such as Haemophilus, Streptococcus, Staphylococcus, Veillonella, Prevotella and Achromobacter also make up the core microbiota observed in bronchiectasis [9, 26, 27]. Interestingly, P. aeruginosa and H. influenzae have been described to competitively inhibit each other, which in turn alters the core microbiota in the non-CF bronchiectasis airway [28]. Non-tuberculous mycobacteria (NTM) form another significant group of pathogens colonizing CF and non-CF airways [29,30,31]. Mycobacterium avium complex (MAC) and Mycobacterium abscessus are most frequently isolated in CF [32, 33] with high rates of multi-drug resistance in these species making them notoriously difficult to treat [34]. NTM belonging to the MAC group are also highly prevalent in non-CF bronchiectasis with a female preponderance [35, 36]. This group of organisms are surprisingly poorly associated with disease severity and exacerbations in the non-CF setting when compared to Pseudomonas [37, 38]. In contrast for CF patients, MAC and M. abscessus are often associated with an aggressive and accelerated lung function decline [39,40,41,42]. Interestingly, bacterial populations do not drastically change between stable and exacerbation states in bronchiectasis. However, viral load has been positively correlated with exacerbations in both CF and non-CF bronchiectasis patients. Infection with viruses belonging to the coronavirus, rhinovirus and influenza A/B virus families are frequently detected during exacerbations of bronchiectasis [43,44,45]. Whether the occurrence of such viruses, forming part of the airway ‘virome’ in bronchiectasis, are a cause or consequence of exacerbations remains to be elucidated [43, 46]. Most attention towards understanding the microbiome in bronchiectasis is directed at the bacteriome. Although fungi are frequently isolated from the same airways, the role of the pulmonary mycobiome in the pathogenesis of these disease states remains largely elusive [47,48,49]. Filamentous fungi belonging to the genus Aspergillus are frequently isolated fungal organisms in sputum samples from CF patients [50, 51]. Among the different species of Aspergillus, A. fumigatus is the most common chronic colonizer in CF [47, 52] Allergic bronchopulmonary aspergillosis (ABPA), an allergic Aspergillus-associated disease, is a frequent co-morbidity in CF [53], while Aspergillus colonization and sensitization has also been independently correlated with lung function declines and radiological severity in CF [54,55,56]. Only a single study to date has shown that fungi belonging to the Aspergillus spp. and Candida albicans are also identifiable in the airways of non-CF bronchiectasis patients [57]. Importantly, in a study of severe asthma patients, Aspergillus fumigatus sensitization has also been associated with poorer lung function and an increased incidence of bronchiectasis, a likely cause and consequence for this anatomical airway distortion [58, 59]. Among yeasts, Candida spp. are frequent colonizers of the bronchiectatic airways [47, 57, 60]. Isolation of Candida albicans from such airways is shown to be a predictor for frequent hospital-exacerbations and declines in lung function [61]. Compared to bacteria, our present understanding of fungal pathogenesis in the context of both CF and non-CF bronchiectasis remains limited and further work is required to determine their prevalence, colonization frequency, host-pathogen interaction and risk factor profile in this key patient group.
Immunology & inflammation
Neutrophil-dominant inflammation is a key feature of bronchiectasis. Sputum neutrophils are higher in bronchiectasis patients versus healthy controls and this correlates with an increased disease severity [62,63,64]. Both interleukin-8 (IL-8) and leukotriene-B4 (LTB4) are key chemo-attractants required for migration and infiltration of neutrophils into bronchiectatic airways [65]. High systemic IL-8 levels are detectable in individuals with bronchiectasis [66,67,68]. Antibacterial neutrophil responses (such as reactive oxygen species (ROS) formation) are activated through the IL-8-CXCR1 axis, but proteolytic cleavage mediated by neutrophil elastase (NE), which itself is associated with exacerbations and lung function decline in bronchiectasis, impairs antibacterial neutrophil functions [69, 70]. Uncontrolled NE activity, as found in CF airways, causes further respiratory tissue damage through degradation of extracellular proteins (such as surfactant proteins [71,72,73]) and cellular surface receptors (such as complement receptors [74]); high NE levels correlating with disease severity and poorer lung function are described in both CF and non-CF bronchiectasis settings [75, 76]. In this context, CXCR receptor antagonists are hypothesized to inhibit neutrophil airway influx and have been shown to be effective in modulating the inflammatory state in bronchiectasis [77, 78]. Airway neutrophils in CF illustrate an impaired phagocytic ability [79]. This is in line with the observation that CF neutrophils have an impaired ROS production, a critical mediator of antimicrobial host defense [80]. Neutrophils defective in their oxidative abilities obtained from non-CF bronchiectasis patients were poorer at bacterial killing when compared to those of healthy controls [81]. Serine proteases are also important neutrophil derived products, released in response to TNF-α signalling. They degrade proteoglycans in the respiratory epithelium subsequently inducing airway damage [82]. In bronchiectasis, activated airway neutrophils secrete an abundance of human neutrophil peptides (HNPs), which have been described to inhibit their phagocytic ability. Importantly, high concentrations of HNPs are detected in both CF and non-CF airways, which in turn may contribute to the decreased phagocytic abilities and higher rates of infection described in both conditions [83]. Poorer clearance of neutrophils by alveolar macrophages further augments the inflammatory state in bronchiectasis [63]. Eosinophils contribute to tissue injury in CF and the presence of eosinophil cationic protein (ECP) heralds the cell activation state. ECP levels are elevated both in the airway and systemically in bronchiectasis [84,85,86]. Other eosinophilic markers including eosinophil protein X and peroxidase follow a similar pattern and like ECP contribute to poorer pulmonary function [87]. Importantly, eosinophilic granule release in CF may be triggered by NE illustrating the cross-granulocyte talk that occurs in the setting of bronchiectasis [88]. T-cells constitute another key component of the inflammatory response in bronchiectasis [89]. In CF, high T-helper 2 (Th2) [90, 91] and Th17 [91] responses are observed. Th2 cytokines such as IL-4, − 13 and TARC/CCL17 are correlated with a decreased pulmonary function in CF-Pseudomonas-colonized patients. Th17 cells, neutrophils and NKT cells are found in abundance in all-cause bronchiectasis compared to healthy controls [92]. While high Th17 infiltrates independently associate with poorer lung function in CF [93], activation of Th17 antigen-specific pathways have been described in non-CF bronchiectasis [94]. IL-17, a central mediator of the Th17 pathway lacks correlation with bronchiectasis disease phenotypes suggestive of the more prominent role that neutrophil-mediated inflammation likely plays in the pathogenesis of bronchiectasis [94]. Both CD8+ T cells and NKT that express pro-inflammatory IFN-γ and TNF-α have been described in paediatric bronchiectasis [95]. Common pro-inflammatory markers such as TNF-α, IL-8, NE and matrix metalloproteinases − 2, − 8 and − 9 (MMP2, MMP8 and MMP9), are all elevated in bronchiectasis with the latter two indicative of a poorer prognostic outcome [96,97,98,99,100]. A seminal study in CF children identified the key risk factors for bronchiectasis: Sly et al. (2013) showed that elevated airway neutrophil elastase activity was major risk factor and predicted bronchiectasis development [101, 102]. Bacterial load in non-CF bronchiectasis has been correlated with increases in airway (NE, IL-8, IL-1β and TNF-α) and systemic (ICAM-1, E-selectin) derived inflammatory markers, phenomena confirmed in vitro using bronchial epithelial cell lines treated with sputum from bronchiectasis patients [103, 104]. Exacerbations of both CF and non-CF bronchiectasis increases inflammation irrespective of bacterial, viral or fungal causation [43, 105, 106]. Interestingly, sTREM-1 a novel inflammatory marker described in a variety of pulmonary disease states including COPD, has also been identified in children with both CF- and HIV-related bronchiectasis although concentrations in the latter setting are highest. High sTREM-1 levels correlate closely with lung function decline and future studies should explore sTREM-1 levels in bronchiectasis of other aetiologies to better understand its role in the pathogenesis of bronchiectasis [107]. Vitamin-D deficiency, observed in CF [108, 109] is associated with increased bacterial infection, exacerbations and poorer lung function [110,111,112]. This is corroborated in non-CF bronchiectasis where it indicates disease severity and associates with more infection, bacterial colonization, airway inflammation and consequently frequent exacerbations [113].
Clinical characteristics and management of bronchiectasis
Bronchietasis patients are clinically characterized by sputum production (upon exercise or spontaneously) leading to productive coughing with mucopurulent masses of yellowish, greenish or brown sputum in the morning or over the day. However, bronchiectasis are mainly detected at time points when irreversible structural damage has already been done to the airway architecture. Bronchiectasis initially may be reversible in children, later probably not. Major genetic diseases associated with bronchiectasis include CF, primary ciliary dyskinesia (PCD, Kartagener syndrome), alpha 1-antitrypsin deficiency, primary immunodeficiencies or other rare disorders like Williams-Campbell syndrome and Marfan syndrome. Major acquired causes are severe bacterial infections (Tuberculosis, Staphylococcus, Klebsiella and others) or postinfectious bronchiolitis obliterans. Notably, also fungal infections can lead to bronchiectasis, particularly ABPA, as a chronic Th2-driven Aspergillus fumigatus-caused pulmonary condition. Based on this, it is essential in the clinical work-up of patients featuring bronchiectasis to screen for these congenital and acquired conditions in order to tailor appropriate treatments and to attenuate disease progression. In a preventive manner, it is key in the above mentioned conditions to diagnose and monitor for pulmonary symptoms and structural changes (using pulmonary function testing and high-resolution computed tomography, HRCT) to avoid established bronchiectasis-related disease. To this end, it is helpful to follow a concept that has been introduced previously to classify forms of bronchitis in children [114,115,116]. An acute bronchitis, usually triggered by a viral infection, resolves within days or one to two weeks. Sometimes – for many reasons of which most are unknown – symptoms do not resolve spontaneously, but persist. This state is called protracted bacterial bronchitis (PBB). While PBB has been initially established for pediatrics, current publications have discussed and transferred this concept to adults {Birring, 2015 #16382;Gibson, 2010 #16381;Martin, 2015 #16380}. PBB is further differentiated into various forms, depending on the tools used to diagnose it [114, 115]. PBB can be further characterized based on different stratifiers:
-
PBB-microbiologic (“PBB-micro”): (1) presence of chronic wet cough (> 4 weeks), (2) respiratory bacterial pathogens growing in sputum or BAL at density of a single bacterial specifies > 104 colony-forming units/ml, and (3) cough resolves following a 2-week course of an appropriate oral antibiotic (usually amoxicillin-clavulanate)
-
PBB-clinical: (1) presence of chronic wet cough (> 4 weeks), (2) absence of symptoms or signs of other causes of wet or productive cough, (3) cough resolves following a 2-week course of an appropriate oral antibiotic (usually amoxicillin-clavulanate)
-
PBB-extended: as above, but cough resolves only after 4 weeks of antibiotics
-
PBB-recurrent: > 3 episodes of PBB per year
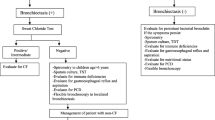
Based on this concept, it is believed that, if left untreated, a fraction of PBBs will progress to chronic suppurative lung disease (CSLD) with radiologically confirmed bronchiectasis (Fig. 1). CSLD differs from bronchiectasis only by lacking the radiographic signs of bronchiectasis on HRCT scans. Clinically, CSLD is diagnosed in children whose chronic wet cough does not resolve with oral antibiotics and in whom other causes are excluded [117, 118]. Although not proven formally for all causes of bronchiectasis, the sequence of progression from PBB over CSLD to bronchiectasis is highly likely, but needs to be substantiated with prospective studies. Of interest is the recent finding that otherwise healthy children with PBB, children with bronchiectasis, and children with CF shared similar core airway microbiota patterns, with H. influenzae making the greatest contribution to the observed similarity, while the microbiota in adults with CF and bronchiectasis were significantly different [25]. The authors concluded that chronic airway infections starts similarly with defective airway clearance, but over time with intervention and host factors, i.e. the underlying cause, progressively diverge.

Model of disease progress towards bronchiectasis in patients with and without CF. Modified from Chang et al. [115]
The prevalence of bronchiectasis in children with CF has recently been assessed in studies conducted by the Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST CF) and others. Although 50–70% of CF patients have CT-defined bronchiectasis by 3 to 5 years of age [119], most young children have isolated, i.e. localized disease, with only the mildest severity of lung abnormalities and lobar disease extent that is well below 50% [120,121,122]. On the other hand, it is clear that once established, bronchiectasis persists and/or progresses despite current optimized standard-of-care therapies in about 75% of young children [121, 122]. Currently great effort is undertaken to close the diagnostic gap from 0 to about 5 years of age, to non-invasively assess the extent of lung disease. The PRAGMA-CF score was developed as a sensitive and reproducible outcome measure for assessing the extent of lung disease in very young children with CF [123]. Moreover, the lung clearance index is a measure of ventilation distribution obtained by the multiple-breath washout technique. Several studies have shown its sensitivity to airway disease in CF and other bronchial diseases [124, 125]. However, in infants with CF, lung clearance index was insensitive to structural disease, as assessed by PRAGMA scoring [126]. In preschool and school-age children with CF’s lung clearance index correlated with total disease extent. Of interest, it had good positive predictive value of about 85%, but a poor negative predictive value of 55% to detect bronchiectasis. Therefore, lung clearance index may be a good surveillance tool to monitor structural lung disease up to school-age in CF [126]. In an effort to identify the preceding-stages of bronchiectasis in CF children by using at least four consecutive biennial volumetric CTs, areas with bronchiectasis on CT scans were marked, further analyzed and associated to potential pre-stages, which were mucus plugging (18%), airway wall thickening (2%) or atelectasis/consolidation in 1% [127].
The basic clinical management of bronchiectasis includes tailored antimicrobial therapy and airway clearance techniques. The latter include mucolytics, such as hypertonic saline and rhDNA, as well as chest physiotherapy and vigorous physical sporting activities. In PBB, oral antibiotics for 2 weeks up to several months have been described to be helpful. Antibiotics commonly used in the clinics include amoxicillin, amoxicillin-clavulanate or second generation cephalosporins. Particularly in patients with CF, gram-negative organisms are treated with inhaled tobramycin, colistin, actreonam or levofloxacin as well as oral inhibitors of gyrases, i.e. ciprofloxacin. The duration of treatment should be guided by symptoms; goal is a symptom-free patient. This can be mostly achieved in young children or patients in the stages of PBB, CSLD and the early stages of bronchiectasis. More specific treatment strategies in bronchiectasis depend on the underlying etiology and include protein augmentation (alpha 1-antitrypsin deficiency), anti-allergic approaches (asthma/ABPA) and/or immunoglobulin substitution (immunodeficiencies).
Imaging of bronchiectasis in CF lung disease
Detection and characterization of bronchiectasis are the domain of thin-section computed tomography (CT). High-resolution CT (HRCT) with 0.6 to 1.5 mm slice thickness serves as a reference standard for imaging. However, pulmonary MRI has gained interest due to the possibility of functional imaging without radiation burden. Moreover, new technical developments overcome the limitations of low MR-signal and low spatial resolution. In CF, standardized reporting using scores or automated quantification are essential requisites to measure and track findings, particularly when results focus on risk stratification. In this context, bronchiectasis is one of the important imaging markers and generally correlates with clinical outcome.
Imaging characteristics of bronchiectasis
Bronchiectasis is defined as irreversible dilation of bronchi in cylindrical, varicose, or a more cystic morphological appearance. In CF, it is often associated with mucus plugging, bronchial wall thickening, and small airway disease [128, 129]. The radiological evaluation of bronchiectasis is based on definition published in the terms for thoracic imaging of the Fleischner Society [130]: “Morphologic criteria on thin-section CT scans include bronchial dilatation with respect to the accompanying pulmonary artery (signet ring sign), lack of tapering of bronchi, and identification of bronchi within 1 cm of the pleural surface.” The so-called signet ring sign is the primary sign for bronchiectasis representing a ring-shaped opacity, whereas the smaller adjacent artery stays for the signet. According to this concept, the extent of bronchial dilation can be quantified using the ratio between bronchi and vessels [128], an approach challenged by a recent pediatric study [131]. On HRCT, the bronchial tree is only visible up to the 6-8th generation [130]. CT findings like the tree-in-bud sign and centrilobular opacity are linked to small airway disease with dilation and inflammation of the ronchiole or mucus plugging in its periphery (Fig. 2) [130]. There are differences in CF bronchiectasis depending on pancreas insufficiency (PI), with PI patients illustrating more severe bronchiectasis [132]. Primary ciliary dyskinesia (PCD) patients have similar CT scores as pancreatic-sufficient (PS) CF patients, but in contrast to CF, no correlation between structural change and clinical parameters has been detected in a previous study [133]. However, recent studies in adult PCD patient cohorts indicate that CT findings relate to lung function changes [134, 135]. There are no clear identifiers of pre-bronchiectasis in imaging. However, mucus plugging has been shown to be a common precursor in CF [127].

15 y/o male, CF-patient, FEV1 predicted 45%. Thin-section HR-reconstruction from MDCT (effective dose of 1.5 mSv). Severe bronchiectasis is visible. Note also dilated bronchi within the periphery of the lung. Air trapping is noted, only in central parenchyma, the CT attenuation appears normal (asterisks)
Imaging can illustrate lung damage even when lung function (such as forced expiratory volume in 1 s, FEV1) is normal [128, 136, 137] (Fig. 3). In contrast to imaging, pulmonary function tests (PFT) are challenging in young children. A complementary role with lung clearance index (LCI) has been described [138]. Regarding the evaluation of the presence and extent of bronchiectasis, CT imaging is accepted as the most sensitive and reproducible modality to date. Using new generation dual source multidetector CT (MDCT) maintaining sub-second acquisition of the whole lung breathing and pulsation artifacts are negligible even in young children and no sedation is needed [139, 140]. Finally, using spectral beam shaping and iterative reconstruction algorithms, ultralow-dose pediatric chest CT can be realized with an effective dose below 0.3 mSv [139]. These conditions, therefore, challenge the routine use of MRI. On the other hand, there are a couple of reasons supporting MRI. The radiation burden of routinely performed volumetric chest CT may be many times higher than that of a third generation of dual source CT recently published. Incremental HRCT with significant gaps lowers the dose but also diagnostic performance and leads to more motion artifact in the pediatric population [141]. Estimated risk of radiation-induced cancer from a pediatric chest CT is small but not negligible, particularly in cases of repeated exposure [142, 143]. MRI has no side effects by radiation enabling long-term surveillance of lung damage. The overall diagnostic performance by scores evaluation of MRI in direct comparison to CT is good to excellent [144,145,146] (Fig. 4). Moreover, apart from polarized 3+ Helium imaging, functional imaging can be easily implemented using perfusion or ventilation weighted standard proton MRI that assesses small airway disease [146,147,148,149] (Fig. 5).

6 y/o female CF-patient, FEV1 predicted 105%. Thin-section HR-reconstruction from MDCT (effective dose of 1 mSv). Mild bronchiectasis, bronchial wall thickening (arrows) and mosaic attenuation (asterisk) are visible

29 y/o male CF-patient, FEV1 predicted 67%. Left side CT, right side MRI at the same day. Upper row, transverse thin-section images from 3D acquisition in breath hold (CT and MRI). Note, despite lower resolution and signal to noise a similar depiction of bronchiectasis is possible. Lower row, expiration images (transverse CT and saggital MRI). In both modalities focal air trapping within the same lung region is demonstrated (arrow heads)

Perfusion map of same patient as in fig. 4 using the noninvasive arterial-spin-labeling technique without contrast media application. The relevant perfusion differences between upper and lower lung regions correlate with morphological damage and air trapping
Clinical value
Standardized reporting of cross-sectional imaging by scoring systems is suitable for several reasons: (a) to assess and to quantify individual progression of lung damage in comparison or complementary to lung function testing, (b) to use total or partial score values as endpoints for interventional studies, and (c) to establish predictive imaging biomarkers. Most CT scoring systems use a semiquantitative scale for the extent and severity of specific findings either based on lobe, involved bronchopulmonary segments, or using an overlay grid [128, 129, 150]. According to the disease-specific prevalence from the imaging abnormalities, sub-scores for bronchiectasis and bronchial wall thickening are more heavily weighted (1, 2). Inter-observer and intra-observer agreement of the common CT scores has shown to be good to excellent [128, 129]. Comparable reproducibility has also been found for MRI using CT or MR-specific adapted scoring systems in small numbers of studies [144, 148]. For semiautomatic evaluation of bronchi dimension excellent inter-observer agreement has been found particularly for the bronchial lumen [151]. On the other hand, mucus can obscure or mimic bronchial wall thickening. Apart from reproducibility, the weighting of abnormalities within the scores, and validation are even more challenging and related to the purpose and the use of score (e.g. interventional or clinical study). As aforementioned imaging is more sensitive than FEV1 particularly in mild disease, and, in assessment of disease progression [128, 136, 137, 152]. In this context scoring of bronchiectasis particularly in the lung periphery is important [137], whereas air trapping, mosaic perfusion, and mucus plugging seem to be more sensitive markers than CT or MRI which detect effects of interventions [153, 154]. The role of bronchiectasis as a robust predictive marker has been shown in several longitudinal observations [150, 155,156,157,158,159]. The extent of bronchiectasis at baseline can predict the number of respiratory tract exacerbations (RTE) [155,156,157,158], and the change of the subscore in a two year follow up is strongly associated with numbers of RTEs where FEV1 did not provide value [156]. This has been similarly described for a decade long observational study [158]. In an older study, a maximum combined score for bronchiectasis and emphysema on HRCT was indicative of a poorer prognosis [159]. In a recent study of patients with severe lung disease awaiting lung transplantation, the combined score of bronchiectasis, bronchial wall thickening, mucus, and consolidation was associated with mortality [150].
Conclusions
Bronchiectasis is a heterogeneous and complex condition and remains a challenge for both diagnostic and therapeutic strategies. While pathomechanisms in the pulmonary compartment share commonalities from a microbiological and immunological perspective, clinical implications and treatment approaches remain challenging and individualized, depending on the underlying disease and the infection status. High-resolution imaging has revolutionized the diagnosis and monitoring of bronchiectasis and will further pave the way for a more precise understanding of disease pathogenesis and treatment response in the future. Therapeutically, lessons learned from the well-known phenotype of CF bronchiectasis are increasingly transferred to the multifaceted genotype and phenotype of non-CF bronchiectasis. Airway infections are treated with inhaled and systemic antibiotics. Mucus clearance can be improved by inhaled therapies and chest physiotherapy, whereas specific anti-inflammatory approaches have still not been clinically established. Prospective future studies are urgently needed to optimize the diagnostic and therapeutic management of bronchiectasis, particularly in children with non-CF bronchiectasis, an indication with a high unmet medical need.
Abbreviations
- ABPA:
-
Allergic bronchopulmonary aspergillosis
- CF:
-
Cystic fibrosis
- COPD:
-
Chronic obstructive pulmonary disease
- CSLD:
-
Chronic suppurative lung disease
- ECP:
-
Eosinophil cationic protein
- FEV1:
-
Forced expiratory volume in 1 s
- HNPs:
-
Human neutrophil peptides
- HRCT:
-
High-resolution computed tomography
- IL-8:
-
Interleukin-8
- LCI:
-
Lung clearance index
- LTB4:
-
Leukotriene-B4
- MAC:
-
Mycobacterium avium complex
- NE:
-
Neutrophil elastase
- PBB:
-
Protracted bacterial bronchitis
- PCD:
-
Primary ciliary dyskinesia
- PFT:
-
Pulmonary function tests
- PI:
-
Pancreas insufficiency
- ROS:
-
Reactive oxygen species
References
Chalmers JD, Aliberti S, Blasi F. Management of bronchiectasis in adults. Eur Respir J. 2015;45(5):1446–62.
Sidhu MK, Mandal P, Hill AT. Bronchiectasis: an update on current pharmacotherapy and future perspectives. Expert Opin Pharmacother. 2014;15(4):505–25.
McShane PJ, Naureckas ET, Tino G, Strek ME. Non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2013;188(6):647–56.
Boyton RJ, Altmann DM. Bronchiectasis: current concepts in pathogenesis, immunology, and microbiology. Annu Rev Pathol. 2016;11:523–54.
Moulton BC, Barker AF. Pathogenesis of bronchiectasis. Clin Chest Med. 2012;33(2):211–7.
Stockley RA. Neutrophils and protease/antiprotease imbalance. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S49–52.
Taggart CC, Greene CM, Carroll TP, O'Neill SJ, McElvaney NG. Elastolytic proteases: inflammation resolution and dysregulation in chronic infective lung disease. Am J Respir Crit Care Med. 2005;171(10):1070–6.
Rab A, Rowe SM, Raju SV, Bebok Z, Matalon S, Collawn JF. Cigarette smoke and CFTR: implications in the pathogenesis of COPD. Am J Physiol Lung Cell Mol Physiol. 2013;305(8):L530–41.
Moran Losada P, Chouvarine P, Dorda M, Hedtfeld S, Mielke S, Schulz A, Wiehlmann L, Tummler B: The cystic fibrosis lower airways microbial metagenome. ERJ Open Res. 2016;2(2):96–115.
Bacci G, Paganin P, Lopez L, Vanni C, Dalmastri C, Cantale C, Daddiego L, Perrotta G, Dolce D, Morelli P et al: Pyrosequencing unveils cystic fibrosis lung microbiome differences associated with a severe lung function decline. 2016, 11(6):e0156807.
King PT, Holdsworth SR, Freezer NJ, Villanueva E, Holmes PW. Microbiologic follow-up study in adult bronchiectasis. Respir Med. 2007;101(8):1633–8.
Kapur N, Grimwood K, Masters IB, Morris PS, Chang AB. Lower airway microbiology and cellularity in children with newly diagnosed non-CF bronchiectasis. Pediatr Pulmonol. 2012;47(3):300–7.
Grimwood K. The pathogenesis of Pseudomonas aeruginosa lung infections in cystic fibrosis. J Paediatr Child Health. 1992;28(1):4–11.
Evans SA, Turner SM, Bosch BJ, Hardy CC, Woodhead MA. Lung function in bronchiectasis: the influence of Pseudomonas aeruginosa. Eur Respir J. 1996;9(8):1601–4.
Davies G, Wells AU, Doffman S, Watanabe S, Wilson R. The effect of Pseudomonas aeruginosa on pulmonary function in patients with bronchiectasis. Eur Respir J. 2006;28(5):974–9.
Hector A, Kirn T, Ralhan A, Graepler-Mainka U, Berenbrinker S, Riethmueller J, Hogardt M, Wagner M, Pfleger A, Autenrieth I, et al. Microbial colonization and lung function in adolescents with cystic fibrosis. J Cystic Fibros. 2016;15(3):340–9.
Konstan MW, Wagener JS, Vandevanter DR, Pasta DJ, Yegin A, Rasouliyan L, Morgan WJ. Risk factors for rate of decline in FEV1 in adults with cystic fibrosis. J Cystic Fibros. 2012;11(5):405–11.
Ren CL, Konstan MW, Yegin A, Rasouliyan L, Trzaskoma B, Morgan WJ, Regelmann W. Multiple antibiotic-resistant Pseudomonas aeruginosa and lung function decline in patients with cystic fibrosis. J Cystic Fibros. 2012;11(4):293–9.
Harun SN, Wainwright C, Klein K, Hennig S: A systematic review of studies examining the rate of lung function decline in patients with cystic fibrosis. Paediatr Respir Rev. 2016;20:55–66.
McDonnell MJ, Jary HR, Perry A, MacFarlane JG, Hester KL, Small T, Molyneux C, Perry JD, Walton KE, De Soyza A. Non cystic fibrosis bronchiectasis: a longitudinal retrospective observational cohort study of Pseudomonas persistence and resistance. Respir Med. 2015;109(6):716–26.
Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol. 2002;34(2):91–100.
Rogers GB, Zain NM, Bruce KD, Burr LD, Chen AC, Rivett DW, McGuckin MA, Serisier DJ. A novel microbiota stratification system predicts future exacerbations in bronchiectasis. Ann Am Thorac Soc. 2014;11(4):496–503.
Chalmers JD, Goeminne P, Aliberti S, McDonnell MJ, Lonni S, Davidson J, Poppelwell L, Salih W, Pesci A, Dupont LJ, et al. The bronchiectasis severity index. An international derivation and validation study. Am J Respir Crit Care Med. 2014;189(5):576–85.
Martinez-Garcia MA, de Gracia J, Vendrell Relat M, Giron RM, Maiz Carro L, de la Rosa CD, Olveira C. Multidimensional approach to non-cystic fibrosis bronchiectasis: the FACED score. Eur Respir J. 2014;43(5):1357–67.
van der Gast CJ, Cuthbertson L, Rogers GB, Pope C, Marsh RL, Redding GJ, Bruce KD, Chang AB, Hoffman LR. Three clinically distinct chronic pediatric airway infections share a common core microbiota. Ann Am Thorac Soc. 2014;11(7):1039–48.
Rogers GB, van der Gast CJ, Cuthbertson L, Thomson SK, Bruce KD, Martin ML, Serisier DJ. Clinical measures of disease in adult non-CF bronchiectasis correlate with airway microbiota composition. Thorax. 2013;68(8):731–7.
Tunney MM, Einarsson GG, Wei L, Drain M, Klem ER, Cardwell C, Ennis M, Boucher RC, Wolfgang MC, Elborn JS. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respir Crit Care Med. 2013;187(10):1118–26.
Rogers GB, van der Gast CJ, Serisier DJ. Predominant pathogen competition and core microbiota divergence in chronic airway infection. Isme j. 2015;9(1):217–25.
Fowler SJ, French J, Screaton NJ, Foweraker J, Condliffe A, Haworth CS, Exley AR, Bilton D. Nontuberculous mycobacteria in bronchiectasis: prevalence and patient characteristics. Eur Respir J. 2006;28(6):1204–10.
Chu H, Zhao L, Xiao H, Zhang Z, Zhang J, Gui T, Gong S, Xu L, Sun X. Prevalence of nontuberculous mycobacteria in patients with bronchiectasis: a meta-analysis. Arch Med Sci. 2014;10(4):661–8.
Viviani L, Harrison MJ, Zolin A, Haworth CS, Floto RA. Epidemiology of nontuberculous mycobacteria (NTM) amongst individuals with cystic fibrosis (CF). J Cystic Fibros. 2016;15(5):619–23.
Roux AL, Catherinot E, Ripoll F, Soismier N, Macheras E, Ravilly S, Bellis G, Vibet MA, Le Roux E, Lemonnier L, et al. Multicenter study of prevalence of nontuberculous mycobacteria in patients with cystic fibrosis in France. J Clin Microbiol. 2009;47(12):4124–8.
Seddon P, Fidler K, Raman S, Wyatt H, Ruiz G, Elston C, Perrin F, Gyi K, Bilton D, Drobniewski F, et al. Prevalence of nontuberculous mycobacteria in cystic fibrosis clinics, United Kingdom, 2009. Emerg Infect Dis. 2013;19(7):1128–30.
Candido PH, Nunes Lde S, Marques EA, Folescu TW, Coelho FS, de Moura VC, da Silva MG, Gomes KM, Lourenco MC, Aguiar FS, et al. Multidrug-resistant nontuberculous mycobacteria isolated from cystic fibrosis patients. J Clin Microbiol. 2014;52(8):2990–7.
Mirsaeidi M, Hadid W, Ericsoussi B, Rodgers D, Sadikot RT. Non-tuberculous mycobacterial disease is common in patients with non-cystic fibrosis bronchiectasis. Int J Infect Dis. 2013;17(11):e1000–4.
Wickremasinghe M, Ozerovitch LJ, Davies G, Wodehouse T, Chadwick MV, Abdallah S, Shah P, Wilson R. Non-tuberculous mycobacteria in patients with bronchiectasis. Thorax. 2005;60(12):1045–51.
Faverio P, Stainer A, Bonaiti G, Zucchetti SC, Simonetta E, Lapadula G, Marruchella A, Gori A, Blasi F, Codecasa L et al: Characterizing Non-Tuberculous Mycobacteria Infection in Bronchiectasis. Int J Mol Sci. 2016;17(11):1913–18.
Maiz L, Giron R, Olveira C, Vendrell M, Nieto R, Martinez-Garcia MA. Prevalence and factors associated with nontuberculous mycobacteria in non-cystic fibrosis bronchiectasis: a multicenter observational study. BMC Infect Dis. 2016;16(1):437.
Esther CR Jr, Esserman DA, Gilligan P, Kerr A, Noone PG. Chronic Mycobacterium abscessus infection and lung function decline in cystic fibrosis. J Cystic Fibros. 2010;9(2):117–23.
Martiniano SL, Nick JA. Nontuberculous mycobacterial infections in cystic fibrosis. Clin Chest Med. 2015;36(1):101–15.
Qvist T, Taylor-Robinson D, Waldmann E, Olesen HV, Hansen CR, Mathiesen IH, Hoiby N, Katzenstein TL, Smyth RL, Diggle PJ, et al. Comparing the harmful effects of nontuberculous mycobacteria and gram negative bacteria on lung function in patients with cystic fibrosis. J Cystic Fibros. 2016;15(3):380–5.
Mussaffi H, Rivlin J, Shalit I, Ephros M, Blau H. Nontuberculous mycobacteria in cystic fibrosis associated with allergic bronchopulmonary aspergillosis and steroid therapy. Eur Respir J. 2005;25(2):324–8.
Gao YH, Guan WJ, Xu G, Lin ZY, Tang Y, Lin ZM, Gao Y, Li HM, Zhong NS, Zhang GJ, et al. The role of viral infection in pulmonary exacerbations of bronchiectasis in adults: a prospective study. Chest. 2015;147(6):1635–43.
Wat D, Gelder C, Hibbitts S, Cafferty F, Bowler I, Pierrepoint M, Evans R, Doull I. The role of respiratory viruses in cystic fibrosis. J Cystic Fibros. 2008;7(4):320–8.
Kieninger E, Singer F, Tapparel C, Alves MP, Latzin P, Tan HL, Bossley C, Casaulta C, Bush A, Davies JC, et al. High rhinovirus burden in lower airways of children with cystic fibrosis. Chest. 2013;143(3):782–90.
Kapur N, Mackay IM, Sloots TP, Masters IB, Chang AB. Respiratory viruses in exacerbations of non-cystic fibrosis bronchiectasis in children. Arch Dis Child. 2014;99(8):749–53.
Chotirmall SH, McElvaney NG. Fungi in the cystic fibrosis lung: bystanders or pathogens? Int J Biochem Cell Biol. 2014;52:161–73.
Moss RB. Fungi in cystic fibrosis and non-cystic fibrosis bronchiectasis. Semin Respir Crit Care Med. 2015;36(2):207–16.
Knutsen AP, Bush RK, Demain JG, Denning DW, Dixit A, Fairs A, Greenberger PA, Kariuki B, Kita H, Kurup VP, et al. Fungi and allergic lower respiratory tract diseases. J Allergy Clin Immunol. 2012;129(2):280–91. quiz 292-283
Sudfeld CR, Dasenbrook EC, Merz WG, Carroll KC, Boyle MP. Prevalence and risk factors for recovery of filamentous fungi in individuals with cystic fibrosis. J Cystic Fibros. 2010;9(2):110–6.
Cimon B, Symoens F, Zouhair R, Chabasse D, Nolard N, Defontaine A, Bouchara JP. Molecular epidemiology of airway colonisation by aspergillus fumigatus in cystic fibrosis patients. J Med Microbiol. 2001;50(4):367–74.
Cimon B, Zouhair R, Symoens F, Carrere J, Chabasse D, Bouchara JP. Aspergillus terreus in a cystic fibrosis clinic: environmental distribution and patient colonization pattern. J Hosp Infect. 2003;53(1):81–2.
Chotirmall SH, Al-Alawi M, Mirkovic B, Lavelle G, Logan PM, Greene CM, McElvaney NG. Aspergillus-associated airway disease, inflammation, and the innate immune response. Biomed Res Int. 2013;2013:723129.
Kraemer R, Delosea N, Ballinari P, Gallati S, Crameri R. Effect of allergic bronchopulmonary aspergillosis on lung function in children with cystic fibrosis. Am J Respir Crit Care Med. 2006;174(11):1211–20.
Wojnarowski C, Eichler I, Gartner C, Gotz M, Renner S, Koller DY, Frischer T. Sensitization to aspergillus fumigatus and lung function in children with cystic fibrosis. Am J Respir Crit Care Med. 1997;155(6):1902–7.
McMahon MA, Chotirmall SH, McCullagh B, Branagan P, McElvaney NG, Logan PM. Radiological abnormalities associated with aspergillus colonization in a cystic fibrosis population. Eur J Radiol. 2012;81(3):e197–202.
Maiz L, Vendrell M, Olveira C, Giron R, Nieto R, Martinez-Garcia MA. Prevalence and factors associated with isolation of aspergillus and Candida from sputum in patients with non-cystic fibrosis bronchiectasis. Respiration. 2015;89(5):396–403.
Menzies D, Holmes L, McCumesky G, Prys-Picard C, Niven R. Aspergillus sensitization is associated with airflow limitation and bronchiectasis in severe asthma. Allergy. 2011;66(5):679–85.
Fairs A, Agbetile J, Hargadon B, Bourne M, Monteiro WR, Brightling CE, Bradding P, Green RH, Mutalithas K, Desai D, et al. IgE sensitization to aspergillus fumigatus is associated with reduced lung function in asthma. Am J Respir Crit Care Med. 2010;182(11):1362–8.
Chotirmall SH, Greene CM, McElvaney NG. Candida species in cystic fibrosis: a road less travelled. Med Mycol. 2010;48(Suppl 1):S114–24.
Chotirmall SH, O'Donoghue E, Bennett K, Gunaratnam C, O'Neill SJ, McElvaney NG. Sputum Candida albicans presages FEV(1) decline and hospital-treated exacerbations in cystic fibrosis. Chest. 2010;138(5):1186–95.
Dente FL, Bilotta M, Bartoli ML, Bacci E, Cianchetti S, Latorre M, Malagrino L, Nieri D, Roggi MA, Vagaggini B, et al. Neutrophilic bronchial inflammation correlates with clinical and functional findings in patients with noncystic fibrosis bronchiectasis. Mediat Inflamm. 2015;2015:642503.
Watt AP, Brown V, Courtney J, Kelly M, Garske L, Elborn JS, Ennis M. Neutrophil apoptosis, proinflammatory mediators and cell counts in bronchiectasis. Thorax. 2004;59(3):231–6.
Wilson CB, Jones PW, O'Leary CJ, Hansell DM, Dowling RB, Cole PJ, Wilson R. Systemic markers of inflammation in stable bronchiectasis. Eur Respir J. 1998;12(4):820–4.
Mikami M, Llewellyn-Jones CG, Bayley D, Hill SL, Stockley RA. The chemotactic activity of sputum from patients with bronchiectasis. Am J Respir Crit Care Med. 1998;157(3 Pt 1):723–8.
Dean TP, Dai Y, Shute JK, Church MK, Warner JO. Interleukin-8 concentrations are elevated in bronchoalveolar lavage, sputum, and sera of children with cystic fibrosis. Pediatr Res. 1993;34(2):159–61.
Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutierrez JP, Hull J, Olinsky A, Phelan EM, Robertson CF, Phelan PD. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med. 1997;156(4 Pt 1):1197–204.
Ayhan G, Tas D, Yilmaz I, Okutan O, Demirer E, Ayten O, Kartaloglu Z. Relation between inflammatory cytokine levels in serum and bronchoalveolar lavage fluid and gene polymorphism in young adult patients with bronchiectasis. J Thorac Dis. 2014;6(6):684–93.
Hartl D, Latzin P, Hordijk P, Marcos V, Rudolph C, Woischnik M, Krauss-Etschmann S, Koller B, Reinhardt D, Roscher AA, et al. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med. 2007;13(12):1423–30.
Chalmers JD, Moffitt KL, Suarez-Cuartin G, Sibila O, Finch S, Furrie E, Dicker A, Wrobel K, Elborn JS, Walker B et al: Neutrophil Elastase Activity is Associated with Exacerbations and Lung Function Decline in Bronchiectasis. American journal of respiratory and critical care medicine 2016.
Hartl D, Griese M. Surfactant protein D in human lung diseases. Eur J Clin Investig. 2006;36(6):423–35.
von Bredow C, Birrer P, Griese M. Surfactant protein a and other bronchoalveolar lavage fluid proteins are altered in cystic fibrosis. Eur Respir J. 2001;17(4):716–22.
von Bredow C, Wiesener A, Griese M. Proteolysis of surfactant protein D by cystic fibrosis relevant proteases. Lung. 2003;181(2):79–88.
Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, Greene C, McElvaney G, Mall M, Doring G. Innate immunity in cystic fibrosis lung disease. J Cystic Fibros. 2012;11(5):363–82.
Tsang KW, Chan K, Ho P, Zheng L, Ooi GC, Ho JC, Lam W. Sputum elastase in steady-state bronchiectasis. Chest. 2000;117(2):420–6.
Mayer-Hamblett N, Aitken ML, Accurso FJ, Kronmal RA, Konstan MW, Burns JL, Sagel SD, Ramsey BW. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J Respir Crit Care Med. 2007;175(8):822–8.
De Soyza A, Pavord I, Elborn JS, Smith D, Wray H, Puu M, Larsson B, Stockley R. A randomised, placebo-controlled study of the CXCR2 antagonist AZD5069 in bronchiectasis. Eur Respir J. 2015;46(4):1021–32.
Planaguma A, Domenech T, Pont M, Calama E, Garcia-Gonzalez V, Lopez R, Auli M, Lopez M, Fonquerna S, Ramos I, et al. Combined anti CXC receptors 1 and 2 therapy is a promising anti-inflammatory treatment for respiratory diseases by reducing neutrophil migration and activation. Pulm Pharmacol Ther. 2015;34:37–45.
Morris MR, Doull IJ, Dewitt S, Hallett MB. Reduced iC3b-mediated phagocytotic capacity of pulmonary neutrophils in cystic fibrosis. Clin Exp Immunol. 2005;142(1):68–75.
Houston N, Stewart N, Smith DS, Bell SC, Champion AC, Reid DW. Sputum neutrophils in cystic fibrosis patients display a reduced respiratory burst. J Cystic Fibros. 2013;12(4):352–62.
King P, Bennett-Wood V, Hutchinson P, Robins-Browne R, Holmes P, Freezer N, Holdsworth S. Bactericidal activity of neutrophils with reduced oxidative burst from adults with bronchiectasis. APMIS. 2009;117(2):133–9.
Shum DK, Chan SC, Ip MS. Neutrophil-mediated degradation of lung proteoglycans: stimulation by tumor necrosis factor-alpha in sputum of patients with bronchiectasis. Am J Respir Crit Care Med. 2000;162(5):1925–31.
Voglis S, Quinn K, Tullis E, Liu M, Henriques M, Zubrinich C, Penuelas O, Chan H, Silverman F, Cherepanov V, et al. Human neutrophil peptides and phagocytic deficiency in bronchiectatic lungs. Am J Respir Crit Care Med. 2009;180(2):159–66.
Koller DY, Gotz M, Eichler I, Urbanek R. Eosinophilic activation in cystic fibrosis. Thorax. 1994;49(5):496–9.
Koller DY, Urbanek R, Gotz M. Increased degranulation of eosinophil and neutrophil granulocytes in cystic fibrosis. Am J Respir Crit Care Med. 1995;152(2):629–33.
Kroegel C, Schuler M, Forster M, Braun R, Grahmann PR. Evidence for eosinophil activation in bronchiectasis unrelated to cystic fibrosis and bronchopulmonary aspergillosis: discrepancy between blood eosinophil counts and serum eosinophil cationic protein levels. Thorax. 1998;53(6):498–500.
Koller DY, Nilsson M, Enander I, Venge P, Eichler I. Serum eosinophil cationic protein, eosinophil protein X and eosinophil peroxidase in relation to pulmonary function in cystic fibrosis. Clin Exp Allergy. 1998;28(2):241–8.
Liu H, Lazarus SC, Caughey GH, Fahy JV. Neutrophil elastase and elastase-rich cystic fibrosis sputum degranulate human eosinophils in vitro. Am J Phys. 1999;276(1 Pt 1):L28–34.
Silva JR, Jones JA, Cole PJ, Poulter LW. The immunological component of the cellular inflammatory infiltrate in bronchiectasis. Thorax. 1989;44(8):668–73.
Hartl D, Griese M, Kappler M, Zissel G, Reinhardt D, Rebhan C, Schendel DJ, Krauss-Etschmann S. Pulmonary T(H)2 response in Pseudomonas aeruginosa-infected patients with cystic fibrosis. J Allergy Clin Immunol. 2006;117(1):204–11.
Tiringer K, Treis A, Fucik P, Gona M, Gruber S, Renner S, Dehlink E, Nachbaur E, Horak F, Jaksch P, et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am J Respir Crit Care Med. 2013;187(6):621–9.
Tan HL, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC. The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med. 2011;184(2):252–8.
Mulcahy EM, Hudson JB, Beggs SA, Reid DW, Roddam LF, Cooley MA. High peripheral blood th17 percent associated with poor lung function in cystic fibrosis. PLoS One. 2015;10(3):e0120912.
Chen AC, Martin ML, Lourie R, Rogers GB, Burr LD, Hasnain SZ, Bowler SD, McGuckin MA, Serisier DJ. Adult non-cystic fibrosis bronchiectasis is characterised by airway luminal Th17 pathway activation. PLoS One. 2015;10(3):e0119325.
Hodge G, Upham JW, Chang AB, Baines KJ, Yerkovich ST, Pizzutto SJ, Hodge S. Increased peripheral blood pro-inflammatory/cytotoxic lymphocytes in children with bronchiectasis. PLoS One. 2015;10(8):e0133695.
Bergin DA, Hurley K, Mehta A, Cox S, Ryan D, O'Neill SJ, Reeves EP, McElvaney NG. Airway inflammatory markers in individuals with cystic fibrosis and non-cystic fibrosis bronchiectasis. J Inflamm Res. 2013;6:1–11.
Guan WJ, Gao YH, Xu G, Lin ZY, Tang Y, Gu YY, Liu GH, Li HM, Chen RC, Zhong NS. Sputum matrix metalloproteinase-8 and -9 and tissue inhibitor of metalloproteinase-1 in bronchiectasis: clinical correlates and prognostic implications. Respirology. 2015;20(7):1073–81.
Guran T, Ersu R, Karadag B, Akpinar IN, Demirel GY, Hekim N, Dagli E. Association between inflammatory markers in induced sputum and clinical characteristics in children with non-cystic fibrosis bronchiectasis. Pediatr Pulmonol. 2007;42(4):362–9.
Patel IS, Vlahos I, Wilkinson TM, Lloyd-Owen SJ, Donaldson GC, Wilks M, Reznek RH, Wedzicha JA. Bronchiectasis, exacerbation indices, and inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170(4):400–7.
Osika E, Cavaillon JM, Chadelat K, Boule M, Fitting C, Tournier G, Clement A. Distinct sputum cytokine profiles in cystic fibrosis and other chronic inflammatory airway disease. Eur Respir J. 1999;14(2):339–46.
Sly PD, Gangell CL, Chen L, Ware RS, Ranganathan S, Mott LS, Murray CP, Stick SM. Risk factors for bronchiectasis in children with cystic fibrosis. N Engl J Med. 2013;368(21):1963–70.
Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, Stick SM, Robinson PJ, Robertson CF, Ranganathan SC. Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med. 2009;180(2):146–52.
Chalmers JD, Smith MP, McHugh BJ, Doherty C, Govan JR, Hill AT. Short- and long-term antibiotic treatment reduces airway and systemic inflammation in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2012;186(7):657–65.
Angrill J, Agusti C, De Celis R, Filella X, Rano A, Elena M, De La Bellacasa JP, Xaubet A, Torres A. Bronchial inflammation and colonization in patients with clinically stable bronchiectasis. Am J Respir Crit Care Med. 2001;164(9):1628–32.
Guan WJ, Gao YH, Xu G, Lin ZY, Tang Y, Li HM, Lin ZM, Jiang M, Zheng JP, Chen RC, et al. Inflammatory responses, spirometry, and quality of life in subjects with bronchiectasis exacerbations. Respir Care. 2015;60(8):1180–9.
Brill SE, Patel AR, Singh R, Mackay AJ, Brown JS, Hurst JR. Lung function, symptoms and inflammation during exacerbations of non-cystic fibrosis bronchiectasis: a prospective observational cohort study. Respir Res. 2015;16:16.
Masekela R, Anderson R, de Boeck K, Vreys M, Steel HC, Olurunju S, Green RJ. Expression of soluble triggering receptor expressed on myeloid cells-1 in childhood CF and non-CF bronchiectasis. Pediatr Pulmonol. 2015;50(4):333–9.
Hall WB, Sparks AA, Aris RM. Vitamin d deficiency in cystic fibrosis. Int J Endocrinol. 2010;2010:218691.
Rovner AJ, Stallings VA, Schall JI, Leonard MB, Zemel BS. Vitamin D insufficiency in children, adolescents, and young adults with cystic fibrosis despite routine oral supplementation. Am J Clin Nutr. 2007;86(6):1694–9.
Simoneau T, Bazzaz O, Sawicki GS, Gordon C. Vitamin D status in children with cystic fibrosis. Associations with inflammation and bacterial colonization. Ann Am Thorac Soc. 2014;11(2):205–10.
Sexauer WP, Hadeh A, Ohman-Strickland PA, Zanni RL, Varlotta L, Holsclaw D, Fiel S, Graff GR, Atlas A, Bisberg D, et al. Vitamin D deficiency is associated with pulmonary dysfunction in cystic fibrosis. J Cystic Fibros. 2015;14(4):497–506.
McCauley LA, Thomas W, Laguna TA, Regelmann WE, Moran A, Polgreen LE. Vitamin D deficiency is associated with pulmonary exacerbations in children with cystic fibrosis. Ann Am Thorac Soc. 2014;11(2):198–204.
Chalmers JD, McHugh BJ, Docherty C, Govan JR, Hill AT. Vitamin-D deficiency is associated with chronic bacterial colonisation and disease severity in bronchiectasis. Thorax. 2013;68(1):39–47.
Wurzel DF, Marchant JM, Yerkovich ST, Upham JW, Petsky HL, Smith-Vaughan H, Masters B, Buntain H, Chang AB. Protracted bacterial bronchitis in children: natural history and risk factors for bronchiectasis. Chest. 2016;150(5):1101–8.
Chang AB, Upham JW, Masters IB, Redding GR, Gibson PG, Marchant JM, Grimwood K. Protracted bacterial bronchitis: the last decade and the road ahead. Pediatr Pulmonol. 2016;51(3):225–42.
Wurzel DF, Marchant JM, Yerkovich ST, Upham JW, Mackay IM, Masters IB, Chang AB. Prospective characterization of protracted bacterial bronchitis in children. Chest. 2014;145(6):1271–8.
Goyal V, Grimwood K, Marchant JM, Masters IB, Chang AB. Paediatric chronic suppurative lung disease: clinical characteristics and outcomes. Eur J Pediatr. 2016;175(8):1077–84.
Goyal V, Grimwood K, Marchant J, Masters IB, Chang AB. Pediatric bronchiectasis: no longer an orphan disease. Pediatr Pulmonol. 2016;51(5):450–69.
Stick SM, Brennan S, Murray C, Douglas T, von Ungern-Sternberg BS, Garratt LW, Gangell CL, De Klerk N, Linnane B, Ranganathan S, et al. Bronchiectasis in infants and preschool children diagnosed with cystic fibrosis after newborn screening. J Pediatr. 2009;155(5):623–U652.
Thia LP, Calder A, Stocks J, Bush A, Owens CM, Wallis C, Young C, Sullivan Y, Wade A, McEwan A, et al. Is chest CT useful in newborn screened infants with cystic fibrosis at 1 year of age? Thorax. 2014;69(4):320–7.
Mott LS, Park J, Gangell CL, de Klerk NH, Sly PD, Murray CP, Stick SM, Australian Respiratory Early Surveillance Team for Cystic Fibrosis Study G. Distribution of early structural lung changes due to cystic fibrosis detected with chest computed tomography. J Pediatr. 2013;163(1):243–8. e241-243
Mott LS, Park J, Murray CP, Gangell CL, de Klerk NH, Robinson PJ, Robertson CF, Ranganathan SC, Sly PD, Stick SM, et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax. 2012;67(6):509–16.
Rosenow T, Oudraad MC, Murray CP, Turkovic L, Kuo W, de Bruijne M, Ranganathan SC, Tiddens HA, Stick SM, Australian respiratory early surveillance team for cystic F. PRAGMA-CF. a quantitative structural lung disease computed tomography outcome in young children with cystic fibrosis. Am J Respir Crit Care Med. 2015;191(10):1158–65.
Kent L, Reix P, Innes JA, Zielen S, Le Bourgeois M, Braggion C, Lever S, Arets HG, Brownlee K, Bradley JM, et al. Lung clearance index: evidence for use in clinical trials in cystic fibrosis. J Cystic Fibros. 2014;13(2):123–38.
Fuchs SI, Gappa M. Lung clearance index: clinical and research applications in children. Paediatr Respir Rev. 2011;12(4):264–70.
Ramsey KA, Rosenow T, Turkovic L, Skoric B, Banton G, Adams AM, Simpson SJ, Murray C, Ranganathan SC, Stick SM, et al. Lung clearance index and structural lung disease on computed tomography in early cystic fibrosis. Am J Respir Crit Care Med. 2016;193(1):60–7.
Tepper LA, Caudri D, Rovira AP, Tiddens HA, de Bruijne M. The development of bronchiectasis on chest computed tomography in children with cystic fibrosis: can pre-stages be identified? Eur Radiol. 2016;26(12):4563–9.
Brody AS, Klein JS, Molina PL, Quan J, Bean JA, Wilmott RW. High-resolution computed tomography in young patients with cystic fibrosis: distribution of abnormalities and correlation with pulmonary function tests. J Pediatr. 2004;145(1):32–8.
de Jong PA, Ottink MD, Robben SG, Lequin MH, Hop WC, Hendriks JJ, Pare PD, Tiddens HA. Pulmonary disease assessment in cystic fibrosis: comparison of CT scoring systems and value of bronchial and arterial dimension measurements. Radiology. 2004;231(2):434–9.
Hansell DM, Bankier AA, MacMahon H, McLoud TC, Muller NL, Remy J. Fleischner society: glossary of terms for thoracic imaging. Radiology. 2008;246(3):697–722.
Kapur N, Masel JP, Watson D, Masters IB, Chang AB. Bronchoarterial ratio on high-resolution CT scan of the chest in children without pulmonary pathology: need to redefine bronchial dilatation. Chest. 2011;139(6):1445–50.
Simanovsky N, Cohen-Cymberknoh M, Shoseyov D, Gileles-Hillel A, Wilschanski M, Kerem E, Hiller N. Differences in the pattern of structural abnormalities on CT scan in patients with cystic fibrosis and pancreatic sufficiency or insufficiency. Chest. 2013;144(1):208–14.
Cohen-Cymberknoh M, Simanovsky N, Hiller N, Gileles Hillel A, Shoseyov D, Kerem E. Differences in disease expression between primary ciliary dyskinesia and cystic fibrosis with and without pancreatic insufficiency. Chest. 2014;145(4):738–44.
Frija-Masson J, Bassinet L, Honore I, Dufeu N, Housset B, Coste A, Papon JF, Escudier E, Burgel PR, Maitre B. Clinical characteristics, functional respiratory decline and follow-up in adult patients with primary ciliary dyskinesia. Thorax. 2017;72(2):154–60.
Shah A, Shoemark A, SJ MN, Bhaludin B, Rogers A, Bilton D, Hansell DM, Wilson R, Loebinger MR. A longitudinal study characterising a large adult primary ciliary dyskinesia population. Eur Respir J. 2016;48(2):441–50.
Bonnel AS, Song SM, Kesavarju K, Newaskar M, Paxton CJ, Bloch DA, Moss RB, Robinson TE. Quantitative air-trapping analysis in children with mild cystic fibrosis lung disease. Pediatr Pulmonol. 2004;38(5):396–405.
de Jong PA, Lindblad A, Rubin L, Hop WC, de Jongste JC, Brink M, Tiddens HA. Progression of lung disease on computed tomography and pulmonary function tests in children and adults with cystic fibrosis. Thorax. 2006;61(1):80–5.
Owens CM, Aurora P, Stanojevic S, Bush A, Wade A, Oliver C, Calder A, Price J, Carr SB, Shankar A, et al. Lung clearance index and HRCT are complementary markers of lung abnormalities in young children with CF. Thorax. 2011;66(6):481–8.
Weis M, Henzler T, Nance JW Jr, Haubenreisser H, Meyer M, Sudarski S, Schoenberg SO, Neff KW, Hagelstein C. Radiation dose comparison between 70 kVp and 100 kVp with spectral beam shaping for non-contrast-enhanced pediatric chest computed tomography: a prospective randomized controlled study. Investig Radiol. 2016;
Tsiflikas I, Thomas C, Ketelsen D, Seitz G, Warmann S, Claussen CD, Schafer JF. High-pitch computed tomography of the lung in pediatric patients: an intraindividual comparison of image quality and radiation dose to conventional 64-MDCT. RoFo. 2014;186(6):585–90.
Bastos M, Lee EY, Strauss KJ, Zurakowski D, Tracy DA, Boiselle PM. Motion artifact on high-resolution CT images of pediatric patients: comparison of volumetric and axial CT methods. AJR Am J Roentgenol. 2009;193(5):1414–8.
Journy NM, Lee C, Harbron RW, McHugh K, Pearce MS, Berrington de Gonzalez A. projected cancer risks potentially related to past, current, and future practices in paediatric CT in the United Kingdom, 1990-2020. Br J Cancer. 2017;116(1):109–16.
Niemann T, Colas L, Roser HW, Santangelo T, Faivre JB, Remy J, Remy-Jardin M, Bremerich J. Estimated risk of radiation-induced cancer from paediatric chest CT: two-year cohort study. Pediatr Radiol. 2015;45(3):329–36.
Roach DJ, Cremillieux Y, Fleck RJ, Brody AS, Serai SD, Szczesniak RD, Kerlakian S, Clancy JP, Woods JC. Ultrashort Echo-time magnetic resonance imaging is a sensitive method for the evaluation of early cystic fibrosis lung disease. Ann Am Thorac Soc. 2016;13(11):1923–31.
Sileo C, Corvol H, Boelle PY, Blondiaux E, Clement A, Ducou Le Pointe H. HRCT and MRI of the lung in children with cystic fibrosis: comparison of different scoring systems. J Cystic Fibros. 2014;13(2):198–204.
Teufel M, Ketelsen D, Fleischer S, Martirosian P, Graebler-Mainka U, Stern M, Claussen CD, Schick F, Schaefer JF. Comparison between high-resolution CT and MRI using a very short echo time in patients with cystic fibrosis with extra focus on mosaic attenuation. Respiration. 2013;86(4):302–11.
Bauman G, Puderbach M, Heimann T, Kopp-Schneider A, Fritzsching E, Mall MA, Eichinger M. Validation of Fourier decomposition MRI with dynamic contrast-enhanced MRI using visual and automated scoring of pulmonary perfusion in young cystic fibrosis patients. Eur J Radiol. 2013;82(12):2371–7.
Eichinger M, Optazaite DE, Kopp-Schneider A, Hintze C, Biederer J, Niemann A, Mall MA, Wielputz MO, Kauczor HU, Puderbach M. Morphologic and functional scoring of cystic fibrosis lung disease using MRI. Eur J Radiol. 2012;81(6):1321–9.
Schraml C, Schwenzer NF, Martirosian P, Boss A, Schick F, Schafer S, Stern M, Claussen CD, Schafer JF. Non-invasive pulmonary perfusion assessment in young patients with cystic fibrosis using an arterial spin labeling MR technique at 1.5 T. Magma. 2012;25(2):155–62.
Loeve M, Hop WC, de Bruijne M, van Hal PT, Robinson P, Aitken ML, Dodd JD, Tiddens HA. Chest computed tomography scores are predictive of survival in patients with cystic fibrosis awaiting lung transplantation. Am J Respir Crit Care Med. 2012;185(10):1096–103.
Martinez TM, Llapur CJ, Williams TH, Coates C, Gunderman R, Cohen MD, Howenstine MS, Saba O, Coxson HO, Tepper RS. High-resolution computed tomography imaging of airway disease in infants with cystic fibrosis. Am J Respir Crit Care Med. 2005;172(9):1133–8.
de Jong PA, Nakano Y, Hop WC, Long FR, Coxson HO, Pare PD, Tiddens HA. Changes in airway dimensions on computed tomography scans of children with cystic fibrosis. Am J Respir Crit Care Med. 2005;172(2):218–24.
Robinson TE, Goris ML, Zhu HJ, Chen X, Bhise P, Sheikh F, Moss RB. Dornase alfa reduces air trapping in children with mild cystic fibrosis lung disease: a quantitative analysis. Chest. 2005;128(4):2327–35.
Wielputz MO, Puderbach M, Kopp-Schneider A, Stahl M, Fritzsching E, Sommerburg O, Ley S, Sumkauskaite M, Biederer J, Kauczor HU, et al. Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am J Respir Crit Care Med. 2014;189(8):956–65.
Bortoluzzi CF, Volpi S, D'Orazio C, Tiddens HA, Loeve M, Tridello G, Assael BM. Bronchiectases at early chest computed tomography in children with cystic fibrosis are associated with increased risk of subsequent pulmonary exacerbations and chronic pseudomonas infection. J Cystic Fibros. 2014;13(5):564–71.
Brody AS, Sucharew H, Campbell JD, Millard SP, Molina PL, Klein JS, Quan J. Computed tomography correlates with pulmonary exacerbations in children with cystic fibrosis. Am J Respir Crit Care Med. 2005;172(9):1128–32.
Loeve M, Gerbrands K, Hop WC, Rosenfeld M, Hartmann IC, Tiddens HA. Bronchiectasis and pulmonary exacerbations in children and young adults with cystic fibrosis. Chest. 2011;140(1):178–85.
Sanders DB, Li Z, Brody AS. Chest computed tomography predicts the frequency of pulmonary exacerbations in children with cystic fibrosis. Ann Am Thorac Soc. 2015;12(1):64–9.
Logan PM, O'Laoide RM, Mulherin D, O'Mahony S, FitzGerald MX, Masterson JB. High resolution computed tomography in cystic fibrosis: correlation with pulmonary function and assessment of prognostic value. Ir J Med Sci. 1996;165(1):27–31.
Funding
This research has been supported by the Singapore Ministry of Health’s National Medical Research Council under its Transition Award (NMRC/TA/0048/2016) (S.H.C). The funding had no impact on the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
JS wrote large parts of the manuscript and created most of the Figs. 2 3 4 and 5. MG, RC and SC wrote major parts of the manuscript and created Fig. 1. DH supervised the whole process and wrote major parts of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Consent for publication has been obtained from all persons for the included images.
Competing interests
Sanjay Chotirmall is a member of the editorial board (Section Editor). Dominik Hartl has an affiliation with University of Tübingen and Roche Basel (I3-DTA, pRED). All other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Schäfer, J., Griese, M., Chandrasekaran, R. et al. Pathogenesis, imaging and clinical characteristics of CF and non-CF bronchiectasis. BMC Pulm Med 18, 79 (2018). https://doi.org/10.1186/s12890-018-0630-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-018-0630-8