
Abstract
Background
Persistent physical symptoms (PPS), also known as medically unexplained symptoms (MUS), affect approximately 50% of patients in secondary care and are often associated with disability, psychological distress and increased health care costs. Cognitive behavioural therapy (CBT) has demonstrated both short- and long-term efficacy with small to medium effect sizes for PPS, with larger treatment effects for specific PPS syndromes, including non-cardiac chest pain, irritable bowel syndrome (IBS) and chronic fatigue syndrome (CFS). Research indicates that PPS conditions share similar cognitive and behavioural responses to symptoms, such as avoidance and unhelpful beliefs. This suggests that a transdiagnostic approach may be beneficial for patients with PPS.
Methods
A randomised controlled trial (RCT) will be conducted to evaluate the efficacy and cost-effectiveness of a transdiagnostic CBT-based intervention for PPS. 322 participants with PPS will be recruited from secondary care clinics. Participants stratified by clinic and disability level will be randomised to CBT plus standard medical care (SMC) versus SMC alone. The intervention consists of 8 CBT sessions delivered by a qualified therapist over a period of 20 weeks. Outcomes will be assessed at 9, 20, 40- and 52-weeks post randomisation. Efficacy will be assessed by examining the difference between arms in the primary outcome Work and Social Adjustment Scale (WSAS) at 52 weeks after randomisation. Secondary outcomes will include mood, symptom severity and clinical global impression at 9, 20, 40 and 52 weeks. Cost-effectiveness will be evaluated by combining measures of health service use, informal care, loss of working hours and financial benefits at 52 weeks.
Discussion
This trial will provide a powered evaluation of the efficacy and cost-effectiveness of a transdiagnostic CBT approach versus SMC for patients with PPS. It will also provide valuable information about potential healthcare pathways for patients with PPS within the National Health Service (NHS).
Trial registration
ClinicalTrials.gov NCT02426788. Registered 27 April 2015. Overall trial status: Ongoing; Recruitment status: No longer recruiting.
Similar content being viewed by others


Background
Medically unexplained symptoms (MUS) refer to persistent bodily symptoms cannot be adequately explained by organic pathology [1]. Although the umbrella term MUS is commonly used in health care, previous literature suggests that patients prefer the term persistent physical symptoms (PPS) [2]. For this reason, the patient-centred label PPS will be used throughout this paper to refer to MUS.
In secondary care, it is estimated that up to 50% of new referrals experience PPS [3]. These symptoms are often associated with profound functional impairment and psychological distress [3, 4]. Approximately 50% of patients with PPS present with co-morbid conditions, including anxiety, depression [3]. Left untreated, the prognosis of PPS patients is poor [1]. PPS are frequently seen in most medical specialties: irritable bowel syndrome (IBS) in gastroenterology, non-cardiac chest pain in cardiology, fibromyalgia (FM) in rheumatology, respiratory cough and breathlessness in respiratory medicine, and functional neurological symptoms in neurology [5]. Large amounts of healthcare and financial benefits go towards the diagnosis and treatment of PPS, with NHS costs amounting to approximately £3 billion per annum in the working population. Output losses due to sickness absence amounts to several billion [6]. Overall quality of life in people with PPS is poor with many not being able to do the things healthy people take for granted.
The management and treatment of patients with PPS is one of the most difficult tasks facing medical specialists in secondary physical healthcare services. An increasing body of evidence shows that cognitive behavioural interventions can be effective in reducing PPS severity and healthcare-related expenditures. Cognitive behavioural therapy (CBT) has been demonstrated to have both short- and long-term efficacy with small to medium effect sizes. Two meta-analyses examined the clinical effectiveness of short-term psychological therapies (including CBT, reattribution training, psychodynamic therapy, hypnosis etc.) for treating patients with PPS. The results indicated a beneficial effect of CBT for PPS [7, 8]. Improvements have also been reported for several specific PPS syndromes, including non-cardiac chest pain [9,10,11], IBS [12, 13], and chronic fatigue syndrome (CFS) [14]. However, more effective treatments undoubtedly need to be developed as effect sizes are at best modest. People with different PPS conditions often share similar cognitive and behavioural responses to symptoms, including fear avoidance beliefs and catastrophic misappraisal [1]. This suggests the possibility of transdiagnostic aetiological factors underlying these disorders, as well as common perpetuating processes and pathways.
There is an accumulating evidence base supporting the efficacy of transdiagnostic approaches for affective disorders. A transdiagnostic approach assumes that similar psychological processes maintain symptoms and disability across conditions. A previous systematic review and meta-analysis concluded that transdiagnostic approaches may be as effective as diagnosis-specific treatments for alleviating anxiety and may even be superior for treating depression. However these conclusions were based on a small number of studies [15]. Norton & Berra (2012) conducted a non-inferiority RCT comparing the efficacy of transdiagnostic CBT to disorder specific CBT for anxiety disorders. The results revealed treatment equivalence between transdiagnostic and disorder specific approaches, providing support for the efficacy of transdiagnostic treatments [16]. Given the large overlap between PPS syndromes, transdiagnostic interventions are needed, which can effectively target these factors to alleviate PPS, increase participation in life and reduce healthcare costs. Fig. 1 is a proposed logic model for this trial, and it illustrates the potential benefits of using a transdiagnostic approach for patients with PPS.

Logic Model of PRINCE Secondary illustrating the potential benefits of using a transdiagnostic approach. MUS, Medically Unexplained Symptoms; NHS, National Health Service; WSAS, Work and Social Adjustment Scale; PHQ-9, Patient Health Questionnaire – 9 item Scale; GAD-7, Generalised Anxiety Disorder – 7 item Scale; PHQ-15, Patient Health Questionnaire – 15 item Scale
This paper presents a study protocol for the PRINCE (Persistent physical symptoms Reduction Intervention: a system Change and Evaluation in secondary care) Secondary trial, which aims to investigate the efficacy and cost-effectiveness of a therapist delivered transdiagnostic cognitive behavioural approach for treating patients with PPS in secondary care. Common transdiagnostic processes will be targeted with a view to bringing about a change in functional impairment and therefore will be measured alongside primary and secondary outcomes. These transdiagnostic process have been previously found to mediate change in chronic fatigue and chronic pain [17,18,19].
Main research question
What is the efficacy and cost-effectiveness of a therapist delivered, transdiagnostic CBT intervention plus standard medical care (SMC) versus SMC alone for the treatment of patients with PPS in secondary medical care?
Research objectives
Primary objectives
-
1.
To assess the efficacy of a therapist delivered, PPS specific (i.e. transdiagnostic) CBT intervention plus SMC for improving daily functioning in patients with PPS compared to SMC alone at 52 weeks after randomisation.
Secondary objectives
-
1.
To estimate whether therapist delivered transdiagnostic CBT plus SMC has a positive impact on symptom severity, mood and self-reported experience of the main symptom compared to SMC alone at 9, 20, 40 and 52 weeks post randomisation.
-
2.
To estimate the cost-effectiveness of therapist delivered transdiagnostic CBT plus SMC versus SMC alone at 26 weeks prior-, as well as 9, 20, 40 and 52 weeks post randomisation.
-
3.
To evaluate patients’ self-rated global measure of change in health resulting from CBT plus SMC versus SMC alone at 9, 20, 40- and 52-weeks post randomisation.
-
4.
To assess changes in psychological distress caused by patient-defined self-rated main problems at 9, 20, 40- and 52-weeks post randomisation.
-
5.
To assess treatment fidelity of the manual-based CBT intervention and to determine the implications for potential rollout in the National Health Service (NHS) if there is evidence of a treatment effect.
-
6.
To investigate putative cognitive and behavioural mediators of change.
Methods
Trial design
A two-arm parallel group randomised controlled trial (RCT).
Method
Three hundred and twenty-two patients with PPS will be individually randomised to CBT plus SMC versus SMC alone. Follow-up assessments will be conducted at 9, 20, 40- and 52-weeks post randomisation.
Setting
This is a multi-centre trial. Treatment will take place at a hospital out-patient department or via telephone. Participants will be recruited from secondary care in London, UK. Clinics include rheumatology, cardiology, respiratory, neurology, gastroenterology, urology or other.
Target population
Inclusion criteria
-
Adults aged 18-65 years with a diagnosis of PPS. PPS is defined as persistent bodily symptoms with no clear cut obvious organic cause [1].
-
Scoring ≥10 on the Work and Social Adjustment Scale (WSAS);
-
Able to read and write in English;
-
Willing to complete all trial visits;
-
Willing and able to give written informed consent.
Exclusion criteria
-
Active psychosis and/or factitious disorder;
-
Headaches as their main and only PPS symptom (given the clinical complexity of differentiating headaches and migraine, headaches will be excluded)
-
Non-epileptic seizures as their main and only PPS symptom. This is due to a large ongoing RCT evaluating a specific cognitive behavioural approach for Dissociative Seizures recruiting from some of the same clinics;
-
Drug or alcohol dependence disorder;
-
Use of benzodiazepines exceeding the equivalence of 10 mg diazepam per day;
-
Being in receipt of or having received CBT interventions for PPS during the past year;
-
Are at imminent risk of self-harm;
-
Participated in PRINCE Primary study (Trial Registration Number: NCT02444520).
Withdrawal criteria
Participants will be withdrawn from the trial if there are any concerns regarding their consent. Participants can decide to withdraw from the study at any point of the trial, without stating a reason. The trial team will be informed if a participant decides to withdraw consent for research follow-up. Patients who discontinue their treatment but do not withdraw from the study will be followed up. Patient withdrawal forms will be used to record potential dropouts and where possible the reason for drop out.
Planned intervention: Transdiagnostic cognitive Behavioural therapy
The intervention will consist of a maximum of 8 one-hour CBT sessions delivered by one of three qualified trial therapists over a period of 22 weeks. Ideally, CBT sessions will be delivered face-to-face every fortnight. Telephone/skype appointments will be offered in exceptional cases (e.g. for patients with mobility issues). The treatment approach will be transdiagnostic and will be flexible to accommodate the needs of the individual and specific issues associated with specific problems. Thus, it will focus on previously identified symptoms that are overlapping across patient groups included in the trial, as well as common cognitive and behavioural responses to symptoms. The treatment will be manualised to aid the standardisation of treatment delivery and facilitate a potential rollout within the NHS and other healthcare systems if found to be effective. CBT sessions will be structured according to four distinct stages: (1) engagement and rationale giving; (2) reducing avoidance by exposure techniques; (3) dealing with symptom-related cognitions and emotions; and (4) relapse prevention. Overall, the intervention aims to help patients to:
-
a)
Develop an understanding of the relationship between cognitive, emotional, physiological and behavioural aspects of their problem,
-
b)
Understand factors that may be maintaining the problem,
-
c)
Learn how to modify behavioural and cognitive responses, which may be maintaining the problem,
-
d)
Engage in avoided activities
-
e)
Address negative thoughts and illness attributions maintaining symptoms,
-
f)
Address emotional dysregulation, anxiety, low mood or low self-esteem, if present,
-
g)
Adopt a healthy sleep routine which often maintains symptoms and disrupts healthy living.
-
h)
Find ways of living with uncertainty
A therapy manual specifically designed for participants will supplement the content of CBT sessions and will act as an aid to the therapist (see Table 1 for a summary). However, the transdiagnostic approach will also be flexible to accommodate specific issues associated with specific problems. For example, participants with IBS may benefit from discussing bowel related problems.
Therapists
Three trained CBT therapists (clinical behavioural therapist or clinical psychologists) will provide CBT sessions. All therapists will receive training prior to delivering the CBT intervention to participants and supervision will be provided throughout the therapy. In case of resignation/parenting leave of a therapist, we will recruit a replacement therapist as quickly as possible. If a CBT session is missed due to therapist planned annual leave, the therapist will attempt to fit in the missing sessions within the five-month treatment period, but no more than one session will take place in any 1 week.
Therapy training
All therapists will have previously been trained in CBT. A half day training will be required for the delivery of the intervention. Thereafter, weekly meetings will be set up at the start to ensure that all therapists are able to conceptualise PPS appropriately. Therapists will be informed of the trial protocol which will include how to deal with any protocol violations and confidential storage of audio-recordings. They will keep records of the therapy sessions in accordance with the guidelines of the clinical service in which they work and in accordance with professional guidelines.
Therapy supervision
All therapists will receive group supervision every month with the CI (TC). During supervision, therapists will have the opportunity to discuss clinical issues that are problematic or challenging. The CI will remain blind to the actual identification of the patient. In addition, the supervision will check that the manual is largely being followed and ensure that the quality of therapy is sustained.
Treatment Fidelity
All therapy sessions will be audio-recorded for treatment fidelity during the trial. A proportion of audio-recordings will be analysed by two independent clinicians once the trial has been completed. A fidelity measure will be developed, which includes overall therapeutic alliance, CBT skills and overall therapist adherence to the manual.
Intervention adherence
The therapist will record how many CBT sessions out of 8 the participants attended, whether they were face-to-face, or telephone consultations and the duration of each session attended. At the end of therapy, the therapist will also score how well the participant adhered to therapy, as well as rate on a session-by-session basis how well the participant adhered to homework tasks.
If participants do not attend a session, the CBT therapist will contact the participant by telephone to ascertain the problem regarding attendance and will discuss options regarding how to proceed. Choices include a telephone session or a re-arranged face-to-face session, so long as the latter is within five working days. Alternatively, the session will be recorded as having not been attended. Telephone sessions will be kept to a minimum and only arranged if circumstances do not allow the patient to attend the face-to-face session.
Standard medical care
Patients in both trial arms will receive SMC. SMC is defined as the continuation of any follow-up consultations as planned with specialised medical staff, including investigations, discussion of the PPS diagnosis, and the prescribing of any medication if required.
Recruitment
Recruitment of patients will be from secondary care outpatient clinics or if recruitment proves difficult GP surgeries will be approached. Medical practitioners will inform potentially eligible patients about the trial and provide them with a trial leaflet.
Study procedure
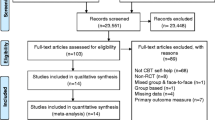
Interested participants will be asked to sign an agreement to be contacted by the research team. The research team will then be responsible for screening patients to check eligibility for the trial as there will be no structured clinical interview prior to inclusion. Eligible patients will then be provided with verbal and written information on the trial to read through. To formally enrol, patients will be required to complete and return a signed informed consent form. Once consent is obtained, they will be asked to complete a baseline questionnaire pack within 1 month of screening. Participants will then be randomised to one of two arms: CBT plus SMC or SMC alone. Outcomes will then be determined at 9, 20, 40 and 52-weeks after randomisation. Fig. 2 provides a CONSORT diagram, outlining the journey of all participants through the trial.

PRINCE Secondary CONSORT Diagram of Study Procedure
Randomisation
Participants will be randomly allocated to one of two trial arms, using a web-based randomisation system managed by the King’s Clinical Trials Unit (KCTU). Randomisation will be at the level of the individual, using block randomisation with random block sizes, stratified by clinic (e.g., cardiology, neurology etc.) and disability level (moderately severe impairment or significant impairment) as indicated by the Work and Social Adjustment Scale.
Treatment allocation will be communicated by email to the trial manager within a period of 24 h. The trial manager will inform the participant of their treatment group by telephone/email that same day. Following allocation to the treatment group the trial manager will inform the therapist, who will contact the participant to arrange the first session.
Proposed sample size
The sample size is based on a previous study: a RCT of rehabilitative treatments for chronic fatigue syndrome [20]. We used data from this trial as it also used the work and social adjustment as an outcome. Based on the White et al. study, we calculated a treatment effect of − 3.6 points on WSAS comparing CBT at 12 months. The within-group SD at 12 months was estimated to be 9.4 points giving a standardised effect size of Cohen’s d = 3.2/9.4 = − 0.38. The sample size calculation (Stata command sampsi) suggests that 161 patients per arm (322 in total) are needed to detect this effect size or a larger one with 90.14% power allowing for a deflation for including baseline measures in the analysis model (factor 0.84 assuming a correlation between baseline and 9-month WSAS of 0.4) and an attrition rate of 25%.
Data management
Data management procedures and blinding will mirror the methods described in a primary care cluster randomised controlled trial [21]. This trial used the same quality assurance methods. In summary, self-report measures and therapy data will be entered onto the InferMed MACRO online database, hosted by the King Clinical Trial Unit (KCTU). This system is used for clinical trials that are managed by the Clinical trials Unit at the Institute of Psychiatry, Psychology and Neuroscience at King’s College London. It follows Good Clinical Practice and FDA 21 CFR Part 11 regulations.
Data entry and management will be undertaken by the research assistants and random checks will be undertaken by the trial manager. The Chief Investigator will be the custodian for the trial data. Patient data will be pseudo-anonymised (allocation of a unique personal identification number (PIN)) after randomisation. Data will be stored on a password-protected computer and in accordance with the General Data Protection Regulation (GDPR). Data will be archived and stored in line with requirements of the sponsor. Paper data will be stored in locked filing cabinets within a locked room and Department.
Blinding
All data will be handled by the research team independent of the clinical team responsible for assessing and treating the patients. As this is a psychotherapy trial it is impossible to blind therapists and patients to which treatment they have been allocated to. Outcomes are all self-report measures and will be collected via post or email. Aside from the trial manger, the trial management team including the chief investigator, statisticians and the independent oversight committees will be blind to treatment allocation. If for any reason unblinding occurs the CI, TSC and DMEC will be notified.
Data collection and follow-ups
Outcome measures will be collected at baseline (pre-randomisation) and at 9, 20, 40, and 52 weeks post-randomisation. Outcome measures will be collected during a time window and will be dependent on the follow-up time-point. Time windows will be as followed; 9 weeks – (time-window 7, 12); 20 weeks – (time-window 18, 35); 40 weeks – (time-window 38, 49); 52 weeks – (time-window 50, 63).
Participants will be asked to complete self-report measures either in writing, electronically via email or over the telephone. If participants decline to complete the full follow-up assessment but have not revoked their consent to be followed up, they will be asked to fill in only the primary outcome questionnaires. Participants who do not return their questionnaires will be contacted by the research worker via telephone/text/email, to remind them to post back their questionnaires or to invite them to complete them over the telephone.
Measures
All measures are summarised in Table 2.
Baseline
-
1.
Demographic Variables: A number of baseline demographic variables will be collected in order to describe the sample. This includes gender, age, ethnicity, occupational status, educational attainment, living arrangements, native language, and the clinic that the patient was recruited from (i.e. cardiology, neurology, rheumatology, respiratory, gastroenterology, urology and other).
-
2.
Clinical information: Patients will be asked to provide clinical information on their symptoms. This includes;
-
◦ Current diagnosis
-
◦ Duration of symptoms
-
◦ Comorbid medical diagnoses
-
◦ Concomitant medication
-
◦ Fibromyalgia assessment: a 4-item measure which assesses whether the participant meets the criteria for fibromyalgia.
-
◦ Health of relative/ close friend: a 4-item measure which assesses the medical history of family and close friends (i.e. heart condition, stroke, neurological condition, condition similar fibromyalgia).
-
◦ The AUDIT alcohol consumption questions (AUDIT-C): a 3-item scale with a range of scores from 0 to 12 measuring alcohol consumption [22].
-
3.
Preferred treatment group: Patients will be asked to indicate their treatment preference post randomisation (i.e. CBT plus SMC, SMC alone, or no preference).
-
4.
Therapist Background Measures: this will include gender, professional background, number of years of experience, full time/part tome status, number of PPS patients treated, highest education level.
Primary outcome measures
-
1.
Functional Impairment: the Work and Social Adjustment Scale (WSAS) is a 5 item scale used to measure the extent to which people’s problems interfere with their ability to carry out normal activities, go to work, partake in private and social activities and impact on relationships [23]. It is a valid and reliable scale which was chosen as it is a routine outcome measure in Increasing Access to Psychological Therapies (IAPT). It has also been used in several other randomised controlled trials evaluating psychological interventions for IBS and CFS [20, 23, 24].
Secondary outcome measures
-
1.
Physical Symptoms: the Patient Health Questionnaire 15 (PHQ15) will be used to measure somatic symptoms [25]. Each item is rated on a 3- point Likert scale (0 = not bothered at all; 1 = bothered a little; 2 = bothered a lot) and the total score can range from 0 to 30 where a higher score indicates higher symptom severity. The PHQ15 is a brief well-validated tool for detecting somatisation [26].
-
2.
Depression: the Patient Health Questionnaire-9 (PHQ-9) will be used to monitor and measure the severity of depression in participants [27]. Each item is rated on a 4-point Likert scale (0 = not at all; 1 = several days; 2 = more than half the days; 3 = nearly every day) and the total score can range from 0 to 27 where a higher score indicates greater depressive severity. The PHQ-9 is a reliable and well-validated measure of depression severity [26].
-
3.
Anxiety: the Generalised Anxiety Disorder – 7 (GAD − 7) questionnaire will be used to measure the severity of GAD in participants [28]. Each item is rated on a 4- point Likert scale (0 = not at all; 1 = several days; 2 = more than half the days; 3 = nearly every day) and the total score can range from 0 to 21 where a higher score indicates greater anxiety. The GAD-7 has demonstrated reliable psychometric properties in the measurement of anxiety in the general population[29].
-
4.
The main presenting symptom: The Persistent Physical Symptom Questionnaire is comprised of three scales to measure (i) severity, (ii) distress and the (iii) problematic nature of the patients main presenting symptom (e.g., chest pain). Each item is scored on a 10-point scale (from 1 = not at all to 10 = extremely). Average scores from the three scales will be used to calculate an overall interference score. This measure was adapted from the Chest Pain questionnaire, which has been previously used for patients with non-cardiac chest pain [30].
-
5.
Global Outcome: the adapted Clinical Global Impression (CGI patient) will be used to measure global change. It has been used in many previous trials of psychosocial treatments [31]. This is rated on a 9-point Likert scale where 1 is completely recovered and 9 is could not get any worse.
-
6.
Costs (Client Service Receipt Inventory): the self-report Client Service Receipt Inventory will be used to assess health service use, informal care, lost work time and financial benefits [32].
-
7.
EuroQoL 5D: the EQ-5D is a reliable and valid tool to measure health related quality of life [33]. Each dimension (mobility, self-care, usual activity, pain/discomfort and anxiety/depression) is rated on 5 levels (1 = no problems; 2 = slight problems; 3 = moderate problems; 4 = severe problems; 5 = extreme problems). The participant will also rate their own perception of their current health on a visual analogue scale ranging from 0 to 100 (0 = the best health you can imagine to 100 = the worst health you can imagine).
Process variables
-
1.
Therapy Process Indicators
-
a.
Treatment attendance and homework log: Therapist will be asked to report
-
i.
Patient attendance
-
ii.
Reasons for session cancellation if cancelled
-
iii.
Mode of delivery (face to face or by telephone)
-
iv.
Length of session
-
v.
Provision of handouts
-
vi.
Percentage of homework completed as rated by the therapist
-
vii.
Number of unplanned telephone calls
-
i.
-
b.
Treatment adherence: Treatment adherence will be rated by the CBT therapist at the end of treatment. The therapist will rate how well the participant adhered to treatment and to what extent they accepted the therapy model. This will be rated on a 4-point Likert scale.
-
c.
Competence rating: a questionnaire completed by the fidelity raters assessing the therapist competence.
-
d.
Satisfaction with treatment (CBT plus SMC only): Satisfaction of treatment will be measured at week 20. Participants will be asked to rate how satisfied they were with the CBT treatment on a 7-point Likert scale (1 Very dissatisfied; 2 Moderately dissatisfied; 3 Slightly dissatisfied; 4 Neither; 5 Slightly satisfied; 6 Moderately satisfied; 7 Very satisfied).
-
e.
Global Improvement (therapists) (CBT plus SMC only): At the end of treatment, CBT therapists will rate how much the participant changed since the start of the study using an adapted version of the Clinical Global Impression (CGI therapist). This will be rated on a 9-point Likert scale where is 1 is completely recovered and 9 is could not get any worse.
-
f.
Self-Rated Patient-Defined Problem (CBT plus SMC only): Participants in the CBT plus SMC group only will be asked to complete the PSYCHLOPS questionnaire. The PSYCHLOPS is a well validated and reliable patient-generated measure [34], which assesses function and well-being on problems that are reported by the participant. As the problems are significant to the participant, it is highly sensitive to change and can be measured throughout the trial. Participants are asked to describe their main problem or problems and how it affects them. All responses to questions are scored.
-
a.
-
2.
Mechanisms of Change: A separate analysis plan will be written for the mediation analysis.
-
a.
The Cognitive Behavioural Responses Questionnaire (CBRQ) is a valid and reliable tool used to assess participants cognitive and behavioural responses to their symptoms [35]. Each item is measured on a five-point Likert scale, scored from 0 (strongly disagree) to 4 (strongly agree) where a higher subscale score indicates more unhelpful cognitions and behaviours.
-
b.
The Acceptance scale is a 9-item subscale measuring pain willingness, taken from the Chronic Pain and Acceptance questionnaire [36] and adapted to focus on willingness to accept symptoms. Participants will be asked to rate each item as its applied to them on a 7-point Likert scale (0 = never true to 6 = always true); where a higher score will indicate greater acceptance.
-
a.
Data collection plan: retention
Retention rates will be monitored and potentially boosted by providing participants with options regarding completion of questionnaires. These will include via post, telephone or email. Thank-you cards will be sent mid trial and end of trial.
Statistical analysis
A statistical analysis plan (SAP) will be developed by the statisticians and agreed with the trial team before database lock. The formal statistical analyses will estimate the difference in mean outcomes between patients randomised to CBT plus SMC and SMC by intention to treat at the various post-treatment observation time points. Estimates of effect sizes together with 95% confidence intervals will be reported. It is planned that the main statistical analysis will use a linear mixed model with maximum likelihood estimation. The data will be analysed on an intention to treat basis.
The linear mixed model will contain post-treatment measures of the primary outcome (at the four follow up time points, i.e. 9, 20, 40 and 52 weeks) as the dependent variables. Fixed effects will consist of:
-
Baseline measures of WSAS;
-
Trial arm;
-
Dummy variables for time points (9, 20, 40 or 52 weeks);
-
Trial arm Χ time interaction terms;
-
Dummy variables for treatment clinic (randomisation stratifier).
-
Dummy variable for disability level (randomisation stratifier)
-
Dummy variables for therapist
Baseline measures of the outcome variable are included as they are known predictors of the outcome and thus should help us to gain precision for effect estimates of interest.
A random effect for participant will be entered into the model to account for correlations between the four repeated measures per participant. A more complex correlation structure (e.g. random intercept and slope model) may be considered if it proved to provide a better model fit.
Standardised effect sizes will be computed to measure the effect of treatment on primary and secondary outcomes at various assessment time points. This will be done by dividing the estimated trial arm difference by the baseline standard deviation of the measure.
We will investigate empirically whether non-adherence with CBT predicts loss to follow-up, and if this were the case, we will use multiple imputation instead of linear mixed modelling to generate inferences that are valid under a realistic missing at random assumption.
We will ensure that patterns of missing data and reasons for missingness are consistent with the CONSORT diagram. Baseline characteristics will be assessed to see if they predict missingness, if so, they will be included in the analysis model. We will check if any post randomisation variables such as compliance are predictive of missingness, and if so this will inform our missing data approach.
Economic evaluation
This analysis aims to compare the 1) service use and costs between CBT + SMC vs SMC, and 2) and assess the cost-effectiveness of CBT + SMC in relation to SMC. Two economic measures will be used to solicit data; i.e., the CSRI and the EQ-5D-5 L. The cost of services will be calculated by combining service use data with relevant unit costs [37,38,39]. Informal care and lost employment will both be valued using average wage rates. The use of services will be described by reporting the number of participants (%) accessing services in each group at different study points and the mean (SD) number of contacts for those using them (i.e. excluding those with zero use). Mean (SD) costs of individual services across the whole sample will also be reported and compared between the two study groups.
Health states data elicited using the EQ-5D-5 L will be combined with population utility weights to derive quality-adjusted-life-years (QALYs) used to measure health benefits in cost-utility analyses [40]. Cost-effectiveness will be assessed by combining the costs data (separately for each perspective) with the WSAS and QALYs. Incremental cost-effectiveness ratios will be computed and uncertainty around the results addressed using cost-effectiveness planes and acceptability curves.
Trial oversight
The trial will be overseen by the Programme Management Group (PMG), as well as two independent committees, the Data Monitoring and Ethics Committee (DMEC) and the Trial Steering Committee (TSC). All committees will be responsible for ensuring that the study is conducted in accordance with the International Conference for Harmonisation of Good Clinical Practice guidelines. The DMEC will oversee the trial data, including serious adverse events and ethics and the TSC will monitor overall progress of the trial and ensure that the study protocol is being adhered to.
Procedures for recording and reporting serious adverse events
Adverse Events (AE) will be assessed at 20, 40 and 52 weeks after randomisation. All serious adverse events (SAE) and reactions (SAR) will be reported immediately to the Chief Investigator, sponsor and DMEC. AE’s that are defined as serious will mirror the criterion described in a primary care cluster randomised controlled trial [21].
In summary, the reporting of SAE’s will be based on the following criterion; (i) death of a participant, (ii) life threatening event (iii) hospitalisation (not including elective hospitalisation for pre-existing condition) (iv) deliberate self-harm or (v) any important medical condition which may influence the participants safety. In addition, an SAE will be reported, if the participant deteriorates in that the level of disability worsens and they are unable.to carry out important daily activities for more than 4 weeks.
All serious adverse events and reactions will be reviewed, by two members of the DMEC without the presence of the CI. If deemed necessary, the scrutinisers would then be unblinded to treatment allocation so that they can then establish whether any serious adverse events were serious adverse reactions to the transdiagnostic approach.
Stopping rules
If deemed necessary, based on either new safety information or lack of recruitment, the trial may be prematurely stopped by the Sponsor, Chief Investigator, DMEC, TSC or Research Ethics Committee (REC). If the study is discontinued, participants enrolled onto the study will be informed and subsequently data collection will stop.
Auditing
This trial will be compliant with the research governance framework and MRC Good Clinical Practice Guideline [41]. The data will be regularly monitored and if requested, access to source data and other documents relating to the trial will be provided to the sponsor and research ethics committee for audit/REC review processes. The chief investigator will supervise a trial manager who will be situated at the Institute of Psychiatry, Psychology and Neuroscience, King’s College London. The trial manager will monitor data collection procedures, including the level of missing data. Furthermore, they will carry out source data verification checks against the paper forms. The trial manager will supervise a research worker to ensure they are fully trained in undertaking data entry/management/cleaning procedures.
The trial statisticians will be affiliated with KCTU and will be responsible for submitting reports to the DMEC and completing the trial Statistical Analysis Plan (SAP). The KCTU SOPs guidelines will be followed which outline the randomisation system, database development and statistics.
Ethics and dissemination
Research ethics approval
South London and Maudsley (SLaM) Hospital have agreed sponsorship. Ethical approval has been granted by the Camberwell St Giles Ethics Committee (Reference 15/LO/0058).
Insurance/indemnity
Standard procedures for insurance of University and NHS employees and sites, and NHS patients will apply.
Dissemination policy
The results will be presented to healthcare professionals nationally and internationally and published in peer-reviewed journals. If the intervention is more effective than the control, we plan to offer training workshops to clinical services within the NHS. We will provide lay summaries to charities and the public via websites who already disseminate information on PPS.
Discussion
PPS are highly prevalent in both primary and secondary care, and are associated with severe physical disability, psychological distress and high health care costs [3, 4, 6]. This paper outlines the study protocol of an RCT designed to evaluate the efficacy and cost-effectiveness of a transdiagnostic cognitive behavioural intervention for adults with PPS in secondary care. The PRINCE Secondary study will be the first trial worldwide to address the efficacy and cost-effectiveness of a manual-based, transdiagnostic, approach for PPS. If it proves to be efficacious, this treatment approach could significantly improve overall functioning in patients with PPS and may lead to substantial long-term economic benefits to the NHS. Moreover, this approach could also be potentially beneficial for treating patients with other debilitating long-term conditions, including diabetes, hypertension and chronic kidney disease.
There are several limitations that need to be considered when assessing the potential impact and implications of our findings. First, we do not have an attention control so any change that occurs in the transdiagnostic CBT group cannot be attributed to the specific contents of the intervention. Second, the present trial protocol deviates from the original protocol. The intention of this study was to recruit a representative sample of therapists to deliver the intervention. However, only three therapists were recruited as opposed to eight which was outlined in the original protocol. This protocol deviation led to the trial becoming an evaluation of the efficacy of CBT delivered by these three therapists. This change in trial objective also led us to recalculate the sample size requirement with the approval of Camberwell St Giles REC.
Availability of data and materials
The protocol does not contain any data. The datasets generated/analysed using this protocol will be anonymised and deposited in a repository after publication. Bona-fide researchers can apply to use the data and materials but are required to clearly specify the research question a priori. No consent was provided for sharing data with third parties.
Change history
12 August 2020
An amendment to this paper has been published and can be accessed via the original article.
Abbreviations
- AE:
-
Adverse event
- BABCP:
-
British Association of Behavioural and Cognitive Psychotherapies
- CBRQ:
-
Cognitive behavioural responses questionnaire
- CBT:
-
Cognitive behavioural therapy
- CFS:
-
Chronic fatigue syndrome
- CGI:
-
Clinical global impression
- CI:
-
Chief investigator
- CSRI:
-
Client service receipt inventory
- DMEC:
-
Data monitoring & ethics committee
- EQ-5D:
-
EuroQoL 5D
- FM:
-
Fibromyalgia
- GAD-7:
-
The generalised anxiety disorder (7-item version)
- GDPR:
-
General data protection regulation
- GP:
-
General practitioner
- IBS:
-
Irritable bowel syndrome
- KCTU:
-
King’s clinical trial unit
- MRC:
-
Medical research council
- MUS:
-
Medically unexplained symptoms
- NHS:
-
National Health Service
- PHQ-15:
-
Patient health questionnaire (15-item version)
- PIN:
-
Personal identification number
- PMG:
-
Programme management group
- PPS:
-
Persistent physical symptoms
- PRINCE:
-
Persistent physical symptoms reduction intervention: a system change and evaluation
- RCT:
-
Randomised controlled trial
- REC:
-
Research ethics committee
- SAE:
-
Serious adverse events
- SAP:
-
Statistical analysis plan
- SAR:
-
Serious adverse reactions
- SMC:
-
Standard medical care
- SOPs:
-
Standard operating procedures
- TM:
-
Trial manager
- TSC:
-
Trial steering committee
- UKFNS:
-
UK Functional Neurological Symptoms
- WSAS:
-
Work & social adjustment scale
References
Deary V, Chalder T, Sharpe M. The cognitive behavioural model of medically unexplained symptoms: a theoretical and empirical review. Clin Psychol Rev. 2007;27(7):781–97.
Picariello F, Ali S, Moss-Morris R, Chalder T. The most popular terms for medically unexplained symptoms: the views of CFS patients. J Psychosom Res. 2015;78(5):420–6.
Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms: an epidemiological study in seven specialities. J Psychosom Res. 2001;51(1):361–7.
Dirkzwager AJ, Verhaak PF. Patients with persistent medically unexplained symptoms in general practice: characteristics and quality of care. BMC Fam Pract. 2007;8(1):33.
Chalder T, Willis C. “Lumping” and “splitting” medically unexplained symptoms: is there a role for a transdiagnostic approach? In: Taylor & Francis; 2017.
Bermingham SL, Cohen A, Hague J, Parsonage M. The cost of somatisation among the working-age population in England for the year 2008-2009. Ment Health Fam Med. 2010;7(2):71–84.
Kleinstäuber M, Witthöft M, Hiller W. Efficacy of short-term psychotherapy for multiple medically unexplained physical symptoms: a meta-analysis. Clin Psychol Rev. 2011;31(1):146–60.
Van Dessel N, Den Boeft M, van der Wouden JC, Kleinstäuber M, Leone SS, Terluin B, Numans ME, van der Horst HE, van Marwijk H. Non-pharmacological interventions for somatoform disorders and medically unexplained physical symptoms (MUPS) in adults. Cochrane Database Syst Rev. 2014;11:CD011142.
Jonsbu E, Dammen T, Morken G, Moum T, Martinsen EW. Short-term cognitive behavioral therapy for non-cardiac chest pain and benign palpitations: a randomized controlled trial. J Psychosom Res. 2011;70(2):117–23.
Kisely SR, Campbell LA, Skerritt P, Yelland MJ. Psychological interventions for symptomatic management of non-specific chest pain in patients with normal coronary anatomy. Cochrane Database Syst Rev. 2010;2010(1):CD004101-004101-CD004101-004147.
Chambers J, Marks E, Russell V, Hunter M. A multidisciplinary, biopsychosocial treatment for non-cardiac chest pain. Int J Clin Pract. 2015;69(9):922–7.
Altayar O, Sharma V, Prokop LJ, Sood A, Murad MH. Psychological therapies in patients with irritable bowel syndrome: a systematic review and meta-analysis of randomized controlled trials. Gastroenterol Res Pract. 2015;2015:549308.
Reme S, Stahl D, Kennedy T, Jones R, Darnley S, Chalder T. Mediators of change in cognitive behaviour therapy and mebeverine for irritable bowel syndrome. Psychol Med. 2011;41(12):2669–79.
Castell BD, Kazantzis N, Moss-Morris RE. Cognitive Behavioral Therapy and Graded Exercise for Chronic Fatigue Syndrome: A Meta-Analysis. Clin Psychol Sci Prac. 2011;18(4):311–24.
Newby JM, McKinnon A, Kuyken W, Gilbody S, Dalgleish T. Systematic review and meta-analysis of transdiagnostic psychological treatments for anxiety and depressive disorders in adulthood. Clin Psychol Rev. 2015;40:91–110.
Norton PJ, Barrera TL. Transdiagnostic versus diagnosis-specific CBT for anxiety disorders: a preliminary randomized controlled noninferiority trial. Depress Anxiety. 2012;29(10):874–82.
Chalder T, Goldsmith KA, White PD, Sharpe M, Pickles AR. Rehabilitative therapies for chronic fatigue syndrome: a secondary mediation analysis of the PACE trial. Lancet Psychiatry. 2015;2(2):141–52.
Turner JA, Holtzman S, Mancl L. Mediators, moderators, and predictors of therapeutic change in cognitive-behavioral therapy for chronic pain. Pain. 2007;127(3):276–86.
Veehof MM, Oskam MJ, Schreurs KM, Bohlmeijer ET. Acceptance-based interventions for the treatment of chronic pain: a systematic review and meta-analysis. Pain. 2011;152(3):533–42.
White PD, Goldsmith KA, Johnson AL, Potts L, Walwyn R, DeCesare JC, Baber HL, Burgess M, Clark LV, Cox D. Comparison of adaptive pacing therapy, cognitive behaviour therapy, graded exercise therapy, and specialist medical care for chronic fatigue syndrome (PACE): a randomised trial. Lancet. 2011;377(9768):823–36.
Patel M, et al. Persistent physical symptoms reduction intervention: a system change and evaluation (PRINCE)—integrated GP care for persistent physical symptoms: protocol for a feasibility and cluster randomised waiting list, controlled trial. BMJ Open. 2019;9(7):e025513.
Bush K, Kivlahan DR, McDonell MB, Fihn SD, Bradley KA. The AUDIT alcohol consumption questions (AUDIT-C): an effective brief screening test for problem drinking. Arch Intern Med. 1998;158(16):1789–95.
Mundt JC, Marks IM, Shear MK, Greist JM. The work and social adjustment scale: a simple measure of impairment in functioning. Br J Psychiatry. 2002;180(5):461–4.
Kennedy TM, Chalder T, McCrone P, Darnley S, Knapp M, Jones R, Wessely S. Cognitive behavioural therapy in addition to antispasmodic therapy for irritable bowel syndrome in primary care: randomised controlled trial. Health Technol Assess Southampton. 2006;10(19):iii–iv, ix-x, 1-67.
Kroenke K, Spitzer RL, Williams JB. The PHQ-15: validity of a new measure for evaluating the severity of somatic symptoms. Psychosom Med. 2002;64(2):258–66.
Kroenke K, Spitzer RL, Williams JB, Löwe B. The patient health questionnaire somatic, anxiety, and depressive symptom scales: a systematic review. Gen Hosp Psychiatry. 2010;32(4):345–59.
Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606–13.
Spitzer RL, Kroenke K, Williams JB, Löwe B. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166(10):1092–7.
Löwe B, et al. Validation and standardization of the generalized anxiety disorder screener (GAD-7) in the general population. Med Care. 2008;46(3):266–74.
Marks E, Chambers J, Russell V, Bryan L, Hunter M. The rapid access chest pain clinic: unmet distress and disability. QJM. 2014;107(6):429–34.
Guy W. ECDEU assessment manual for psychopharmacology. Rockville: National Institute of Mental Health; 1976. p. 218–22.
Beecham J, Knapp M. Costing psychiatric interventions. Measuring Mental Health Needs. 2001;2:200–24.
Brooks R. EuroQol: the current state of play. Health policy. 1996;37(1):53–72.
Ashworth M, et al. Measuring mental health outcomes in primary care: the psychometric properties of a new patient-generated outcome measure,'PSYCHLOPS'(‘psychological outcome profiles’). Primary Care Mental Health. 2005;3:261–70.
Ryan EG, Vitoratou S, Goldsmith KA, Chalder T. Psychometric properties and factor structure of a long and shortened version of the cognitive and Behavioural responses questionnaire. Psychosom Med. 2018;80(2):230–7.
McCracken LM, Vowles KE, Eccleston C. Acceptance of chronic pain: component analysis and a revised assessment method. Pain. 2004;107(1-2):159–66.
National Schedule of Reference costs 2017/18. In. : NHS Improvement 2018; 2018. https://improvement.nhs.uk/documents/1972/1__Reference_costs_201718.pdf.
BNF 77 (British National Formulary) March - September 2019: BMJ Publishing group ltd and Royal Pharmaceutical Society; March 2019.
Curtis L, Burns A. Unit Costs of Health and Social Care 2018, Personal Social Services Research Unit. Canterbury: University of Kent; 2018.
Devlin NJ, Shah KK, Feng Y, Mulhern B, van Hout B. Valuing health-related quality of life: an EQ-5D-5 L value set for England. Health Econ. 2018;27(1):7–22.
MRC guidelines for management of global health trials. https://mrc.ukri.org/documents/pdf/guidelines-for-management-of-global-health-trials/. 2017.
Acknowledgements
PRINCE research staff: Alisia Carnemolla, Nicola Ferreira, Jennifer Robertson, Shinal Patel, Paige Fisher-Smith, Abigale Childs and Richard Turner,
PRINCE trial CBT therapists: David McCormack, Claire Willis and Fabio Simiao.
DMEC: Duncan Dymond (chair), Helen Poole, Anisur Raman, Rebecca Walwyn.
TSC: Alan Carson (chair), Niels Detert, Rebecca Lu, Ruth Pickering.
Patient Representative: Lindsay Gibbs.
Consultants: Dr. Tom Britton, Dr. Biba Stanton, Dr. Nora Ng, Professor Andy Cope, Dr. Sujith Subesinghe, Dr. Sardar Bahadur, Dr. Guy Chung-Faye, Dr. Sukh Chatu, Dr. Pathmanathan, Professor Bruce Kirkham, Dr. Alexandra Kent, Dr. Surrinder Birring, Dr. Geoffrey Warwick, Dr. Giovanni Sanna, Dr. Luay Zebouni, Dr. Dobrina Hull, Dr. Catherine Hughes, Dr. Patrick Dubois, Dr. Iona Coltart, Dr. Richard Turner. We would also like to acknowledge all other NHS staff involved in the recruitment of PRINCE Secondary.
Sponsor information
King’s College London; South London & Maudsley NHS Foundation Trust.
Funding
The study is funded by Guy’s and St. Thomas’ Charity. This funding source had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data, or decision to submit results.
Author information
Authors and Affiliations
Contributions
TC, RM-M, MA, PMC, MH1, AD, SL, TG were involved in the design of the study and were co-applicants for funding. TC led the grant application and is chief investigator. TC developed the intervention. PMC led on the Health Economic evaluation. SL and KJ led on statistical analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was granted by the Camberwell St Giles Research Ethics Committee, REC reference number 15/LO/0058. Written informed consent will be obtained from all participants.
Consent for publication
No details, images or videos relating to an individual person are intended to be published.
Competing interests
MH reported grants from Innovative Medicines Initiative and European Federation of Pharmaceutical Industries and Associations, outside the submitted work. In addition, TC and RMM declared the following; Organisational financial interests: TC received ad hoc payments for conducting workshops on evidence-based treatments for persistent physical symptoms. TC has received grants from NIHR programme grants, HTA, RfPB, Guy’s and St Thomas Charity, King’s Challenge Fund, Muscular Dystrophy, Multiple Sclerosis Society. KCL received payment from Taylor and Francis for editorial role. RMM currently receives grant funding from NIHR programme grants, Breast Cancer Now, Crohn’s and Colitis UK, and National MS Society. In the previous 36 months, RMM received funding from MS society UK and NIHR HTA. In 2019, payments from Taylor and Francis to KCL for RMM’s role as Editor of Health Psychology Review. Payments for adhoc lectures and workshops on Long term conditions. Personal financial interests: TC is the author of several self-help books on chronic fatigue and received royalties in the past. TC received expenses and ad hoc payment for role as external examiner NUI Galway and Waterford Institute of Technology. TC received expenses for keynote speeches at UK Society for Behavioural Medicine, BABCP Conferences (travel and accommodation). TC received ad hoc payments for conducting workshops on evidence-based treatments for persistent physical symptoms. RMM received payments for her role as National Advisor to NHS England for Increasing Access to Psychological Therapies (IAPT) for People with Long Term Conditions from 2011 to 2016. RMM received ad hoc payments for workshop training in IBS in 2017 and 2018 and this will continue in 2019. RMM receives ad hoc consultancy payments from Mahana therapeutics and this is likely to continue in 2019. RMM has stock options in Mahana therapeutics. RMM received travel expenses for keynote speech to Internal Society of Behavioural Medicine. In 2019 RMM will be a keynote speaker for Association for Researchers in Psychology and Health (the Netherlands), European Health Psychology Society Annual Conference (Croatia), and the 9th World Congress of Behavioural and Cognitive Therapies (Germany). Travel and accommodation expenses will be reimbursed. All other authors declare no conflicts of interest. The authors acknowledge the financial support of the Department of Health via the National Institute for Health Research (NIHR) Specialist Biomedical Research Centre for Mental Health award to the South London and Maudsley NHS Foundation Trust (SLaM) and the Institute of Psychiatry at King’s College London. The views expressed in this article are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Chalder, T., Patel, M., James, K. et al. Persistent physical symptoms reduction intervention: a system change and evaluation in secondary care (PRINCE secondary) – a CBT-based transdiagnostic approach: study protocol for a randomised controlled trial. BMC Psychiatry 19, 307 (2019). https://doi.org/10.1186/s12888-019-2297-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12888-019-2297-y