
Abstract
Background
Phenotypic difference is general in Mendelian disease. Due to the extremely low incidence for a single disease, phenotype spectrum needs to be expanded. Meanwhile, earlier knowledge says patients who suffered from two kinds of different Mendelian disease are very rare.
Case presentation
We describe a case of neonatal male with genital anomalies, growth delay, skin hyperpigmentation, chronic lung disease with recurrent infection, anemia, and severe deafness. Without any clear etiology after routine workflow, whole exome sequencing was carried on. A pathogenic de novo SAMD9 mutation and compound heterozygous likely-pathogenic variants in SLC19A2 were identified. Some symptoms were improved after the patient was treated with vitamin B1. Unfortunately, the boy died from sepsis and multiple organ failure before 1 year old.
Conclusion
Combining the phenotype and clinical progress of treatment, we report that it is the first case of a patient with both MIRAGE syndrome and TRMA syndrome.
Similar content being viewed by others


Background
Many neonatal patients with suspected genetic causes are not given a clear diagnosis after the workflow of routine clinical practice such as the recognition of phenotypes, imaging studies or biochemical tests even with biopsy investigations. Genetic tests such as karyotyping, chromosome microarray or the targeted gene panel testing are often carried out in a stepwise approach to search single genetics etiological cause, while many patients remain undiagnosed [1]. Undiagnosed patients may miss the potential treatments, as well as, unable to evaluate the recurrence risk in subsequent pregnancies. A costly and lengthy process for resolving such rare and complicate cases, is commonly called “diagnostic odyssey” [2]. It means huge burdens for families and our medical system.
The next-generation sequencing technology has proven to be an effective way in establishing causes for many rare genetic diseases [3, 4]. Mendelian diseases are estimated to occur at a rate of 40 to 82 per 1000 live births [5]. What is more important is that recent studies show that up-to 6% patients have more than one kinds of genetic disorder [5, 6]. With the medical exome test, we can help increase the diagnosis rate to 50% (data not show) in patients with suspicious Mendelian disease. Here we report a boy with pathogenic de novo mutation in SAMD9 and likely-pathogenic, compound heterozygote mutations in SLC19A2. Since newborn, the boy suffered from recurrent infection, anemia and severe deafness. So far, it is the first case of a patient with both MIRAGE syndrome and TRMA syndrome.
Case presentation
Patient
The Chinese Han male patient was born at 31 weeks gestational age by a spontaneous vaginal delivery, with 1000 g ± 0.2 birth weight. At birth, Apgar scores were 7, 9 and 10 at 1 min, 5 min and 10 min respectively. The patient’s mother was 33 years old with polycystic ovarian syndrome. No information regarding the father had been available. Although the entire pregnancy was uneventful without complications, the fetus was found to be IUGR. Right after the birth, the baby presented some dysmorphic features, such as dysphagia, recurrent infection, as well as hyperpigmentation, excessive body hair and thick eyebrows. Some of the facial appearances faded away gradually with growth. Hypoplastic genitalia was noticed to be female like but chromosome test turned out 46, XY. The penis is visible, while testis and scrotum are not. Ultrasound was carried on at 10 months old, which showed the left testicle is in the inguinal canal while the right is in right lower pelvic cavity. The bilateral epididymis was undetected. No solid mass was found in bilateral scrotum.
Recurrent fever occurred since 3 months old, but sometimes, the results of routine blood testing for inflammation, infections and autoimmune diseases were unremarkable and no obvious clinical features of sepsis were observed. Neutropenia and clinical features of sepsis developed during the 6 months. Due to respiratory distress and chronic lung infection, mechanical ventilation was implemented for about 2 months. Escherichia coli, Klebsiella pneumoniae and Stenotrophomonas maltophilia were found separately via sputum culture. Anomalous pulmonary venous drainage was detected at 6 months and surgery was performed, after which nearly normal cardiac function was restored.
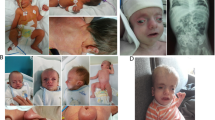
Enteral nutrition was initiated at the third day after birth since the patient has difficultly to suck and swallow. He still suffered from severe growth retardation (less than 3SD) even enteral feeds was given by tube > 150 mL/kg/d and daily calories intake > 130 kcal/kg/d. He had severe malnutrition (weighed 3000 g) and presented chronic diarrhea when enteral feeds were increased. He got severely developmental delay and did not meet the growth milestones at 10 months old (unable to lift his head well or sit alone) (Fig. 1a and b [7]).

Photograph and growth curve of the proband. a Photo of the proband’s back at 2 months, he is extremely thin and skeletonizing. b The proband with low birth weight grew slowly and lagged obviously behind the referent range [7]
Thrombocytopenia and anemia developed since 2 months old and blood transfusion was initiated. Because of matched manifestations such as short status, hyperglycemia, and anemia, vitamin B1 was tried to use to cure the infant. Whilst megaloblastic anemia was partially controlled, granulocytopenia was not.
Since 3 months old, there were recurrent onsets of seizures, no abnormal findings in repeated cerebral spinal fluid for biochemical and pathogen testing. Magnetic resonance imaging showed poor myelination while electroencephalogram did not indicate any remarkable sign. Auditory brainstem response test showed moderate-severe deafness. Blood gas analysis showed no acidosis. Glucose and electrolyte levels were within normal range.
There was no agenesis phenotype in adrenal glands detected by ultrasonography. Plasma cortisol was 18.1 μg/dL (referent range 5-25 μg/dL) while plasma ACTH was 29.7 pmol/L (referent range 0-10.21 pmol/L).
Finally, the patient died from sepsis at 12 months. Recurrent fever, recurrent seizure, malnutrition, developmental delay and chronic diarrhea persisted during the last several months.
Genetic testing
Peripheral blood samples were drawn from the infant and his parents after the informed consent. Genome DNA was prepared with magic beads (Zeesan Biotech, Xiamen, China). Sequencing libraries were prepared with standard TruSeq protocol and followed by liquid phase exome capture and were sequenced with Illumina HiSeq 2000. We analyzed sequencing data with standard practice. Briefly, raw sequencing reads were aligned to GRCh37+ decoy genome with BWA and processed with sambamba view. Variant calling was done with Sentieon and further filtered by Freebayes. Filtered variants were annotated with SnpEff and filtered against population frequency (gnomAD and Euler Genomics in-house Han Chinese population database). Mendelian transmission types of mutations were determined by standard practice. Sanger sequencing was used to verify the de novo SAMD9 mutation by mutant sequence-specific primers (PF: 5’GACTTGACCCAGTGTATCTG3’ and PR: 5’GTCTATC TTCTGCAGTACTC3’).
Filtering against 0.1% population frequency and functional annotation revealed a pair of SLC19A2 (NM_006996) compound heterozygous mutations in the boy (c.1256G > A (p.R419H), rs530420883, paternal origin and c.1052 T > C (p.V351A), rs748588472, maternal origin), as well as a de novo heterozygous missence mutation c.2920 G > A (p. E974K) in SAMD9 gene (NM_017654) (Fig. 2a, b).

Pedigree and verification of the de novo variation in SAMD 9 in the family. a The proband has a heterozygote variant but the parents not. b Pedigree of the patient is shown with SAMD9 / SLC19A2 genotype information. Black square indicates the patient affected by MIRAGE syndrome caused by a de novo variant in SAMD9. Rhombus indicate stillbirth. NA denotes genotype not available. Square filled with dots indicates the proband suffered from TRAM syndrome. The compound heterozygote found in the patient was carried by father and mother respectively
All three variants are not recorded in open databases (HGMD, ClinVar and UniProt). They affect highly conserved amino acid residues (UCSC) and are predicted to be disease causing (mutationtaster), damaging (SIFT) or probably damaging (Polyphen 2). Neither c.1256G > A (p.R419H) nor c.1052 T > C (p.V351A) in SLC19A2 was reported earlier, allele frequencies were around 0.00002 and 0.00009 respectively (gnomAD). Most heterozygotes of c.1052 T > C (p.V351A) were from east Asia showed in gnomAD. The variant was found in both healthy individuals and tumor patients in China from DiseaseDX database (https://diseasedx.virgilbio.com/). The compound heterozygotes in SLC19A2 gene were first reported here in TRMA associated disease.
Additionally, the SAMD9 mutation c.2920 G > A (p.E974K) has been reported as pathogenic in a patient with MIRAGE syndrome previously [8].
Discussion and conclusions
With whole exome sequencing, we found one heterozygous variant in SAMD9 and two compound heterozygous mutants in SLC19A2.
SAMD9 was known to be associated with MIRAGE syndrome, which is a newly reported disease (OMIM ID: 617053) [8, 9]. The core symptoms were adrenal hypoplasia, developmental delay, genital abnormality, anemia, recurrent infection, chronic diarrhea. Reports show the full clinical spectrum of SAMD9 defects still need to be determined [10, 11]. Adrenal hypoplasia has been the most consistent manifestation of the MIRAGE syndrome. The synopsis was seen in 19 out of 21 evaluated patients with MIRAGE syndrome [8, 11, 12]. This fact highlights the significance of our patient’s lacking adrenal features. One patient in Federica Buonocore’s report [12] had no adrenal insufficiency who has a frame shift mutation in SAMD9. Also, one patient in Satoshi Narumi’s research had normal-sized adrenal gland despite a high plasma corticotropin level [8]. The normal ultrasound result for adrenal and higher plasma cortisol reappeared in our patient. SAMD9 is widely expressed and is regulated by TNF-alpha. The protein may play a role in inflammatory response and the control of extra-osseous calcification. Research shows SAMD9 associated abnormality was caused by gain-of-function mutation. In the affected fibroblast cells endosome organization changed [8]. The reduced cell proliferation could be rescued by another loss-of-function mutant in SAMD9 [12]. Maybe there are some other unknown regulated variant in both our patient and the Satoshi Narumi’s patient. Our experience verified the variable expressivity of the adrenal phenotype in MIRAGE syndrome.
c.2920 G > A (p.E974) in SAMD9 has been reported to be pathogenic in a patient with MIRAGE syndrome previously [8]. MIRAGE syndrome matched multiple clinical manifestations in the patient include myelodysplasia, infection, the restriction of growth, genital phenotypes, enteropathy, poor sucking capability, dysphagia, and choking. On the basis of the same mutation and similar synopsis with earlier report [8], we can make the diagnosis of MIRAGE syndrome for the infant. TRMA syndrome is characterized with sensorineural deafness, developmental delay, seizures, megaloblastic anemia, sideroblastic anemia (OMIM ID: 249270)[ 13, 14]. The syndrome is caused by SLC19A2 which encodes Thiamine transporter 1(THTR-1), which is co-expressed with THTR-2 in most tissues except bone marrow, pancreas islet and cochlear cells. THTR-2 acts as a compensate of THTR-1 when transporting thiamine. Pathogenic compound heterozygotes or homozygotes could damage the function of THTR-1, while clinical synopsis mainly occurred in hearing, blood glucose and hematopoiesis [15].
c.1256G > A (p.R419H) and c.1052 T > C (p.V351A) in SLC19A2 are not reported earlier. These two mutations were recorded with allele frequency were 0.00002, 0.00009 in gnomAD and 0, 0.0005 respectively in Chinese (http://diseasedx.Virgi lbio.com/variation/list?id=01f1351e5929ca5c84a545ac59f4027d&table=create1000GenomeDisease&variationType=part1proteins&version=38). p.V351A locates in the transmembrane region of THTR-1 (338th to 354th amino acid). The mutant residue is smaller than the wild-type residue. This size difference can affect the contacts with the lipid-membrane. Thus, change the transmembrane domain. p.R419H is in the cytoplasmic topological domain (410th to 419th amino acid). This variant changed the size of amino acid and the charge and might lead to changing interactions with other molecules or residues [16].
The compound heterozygotes and the associated phenotypes made the diagnosis of TRMA well documented. After supplementation with thiamine for one-month, clinical manifestations of anemia and granulopenia got better. In combination with the gene mutation, we speculated that anemia in the proband might be caused by the SLC19A2 mutations resulting in THTR-1 dysfunction.
There is no report about the relation between SAMD9 and SLC19A2 either their expressed products. The associated clinical synopsis such as myelodysplasia, seizure, anemia, granulopenia and infection may be caused by separating mechanisms.
As a conclusion, we found the patient suffered from both TRMA and MIRAGE syndromes. Coincidences of two Mendelian diseases in one patient are still very rare, but these cases can be found by next-generation sequencing. The medical exome test is a useful tool when face to undiagnosed patients and could be used as an effected alterative in genetics lab.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the [28875-28877] repository, [https://pan.baidu.com/s/1P6c8i9zv5L9JA8tjCAfUfQ/j0k1].
Abbreviations
- HGMD:
-
Human Gene Mutation Database
- IUGR:
-
Intrauterine growth retardation
- MIRAGE:
-
Myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy
- OMIM:
-
Online Mendelian Inheritance in Man
- TRMA:
-
Thiamine-responsive megaloblastic anemia
- UCSC:
-
University of California, Santa Cruz Genome Browser Home
References
Gahl WA, Wise AL, Ashley EA. The undiagnosed diseases network of the national institutes of health: a national extension. JAMA. 2015;314(17):1797–8.
Carmichael N, Tsipis J, Windmueller G, Mandel L, Estrella E. “Is it going to hurt?”: the impact of the diagnostic odyssey on children and their families. J Genet Couns. 2015;24(2):325–35.
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet. 2013;14(10):681–91.
Chen PC, Yin J, Yu HW, Yuan T, Fernandez M, Yung CK, Trinh QM, Peltekova VD, Reid JG, Tworog-Dube E, et al. Next-generation sequencing identifies rare variants associated with Noonan syndrome. Proc Natl Acad Sci U S A. 2014;111(31):11473–8.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369(16):1502–11.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, Xiao R, Ding Y, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376(1):21–31.
Department of Maternal and Child Health and Community Health of the Ministry of Health of the People’s Republic of China Ciop, Research collaboration on the physical development of children in nine cities. Chinese child growth standard and growth curve. Shanghai: The Second Military Medical Uniersity (SMMU) Press; 2009.
Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, Toyoshima K, Tanaka Y, Fukuzawa R, Miyako K, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48(7):792–7.
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore. MIM Number: {617053}: {2016-07-25}: . World Wide Web URL: https://omim.org/. Accessed 14 Jan 2019.
Schwartz JR, Wang S, Ma J, Lamprecht T, Walsh M, Song G, Raimondi SC, Wu G, Walsh MF, McGee RB, et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia. 2017;31(8):1827–30.
Shima H, Koehler K, Nomura Y, Sugimoto K, Satoh A, Ogata T, Fukami M, Juhlen R, Schuelke M, Mohnike K, et al. Two patients with MIRAGE syndrome lacking haematological features: role of somatic second-site reversion SAMD9 mutations. J Med Genet. 2018;55(2):81–5.
Buonocore F, Kuhnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, Khajavi N, Didi M, Brady AF, Blankenstein O, et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest. 2017;127(5):1700–13.
Labay V, Raz T, Baron D, Mandel H, Williams H, Barrett T, Szargel R, McDonald L, Shalata A, Nosaka K, et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet. 1999;22(3):300–4.
Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore. MIM Number: {249270}: {2017-06-08}: . World Wide Web URL: https://omim.org/. Accessed 14 Jan 2019.
Xian X, Liao L, Shu W, Li H, Qin Y, Yan J, Luo J, Lin FQ. A novel mutation of SLC19A2 in a Chinese Zhuang ethnic family with thiamine-responsive megaloblastic anemia. Cell Physiol Biochem. 2018;47(5):1989–97.
Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010;11:548.
Acknowledgments
We are grateful to the patient and his family.
Funding
This work was partly supported by Major Medical Collaboration and Innovation Program of Guangzhou Science Technology and Innovation Commission (grant number 201604020020) to VWZ. Another grant (National Key Research and Development Project, 2018YFC1002600) also supported our work in data analysis. Both the funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
AY planned and designed of the work. YaZ collected the data, drafted and subsequently revised the study. YiZ conducted the key bioinformatics analysis of the data. VWZ finished the next-generation sequencing and took part in the data analysis, also joined the revision of the work. HD conducted sanger sequencing and the interpretation of the data. CZ collected the clinical information of the patient. All authors read and approved the final manuscript.
Authors’ information
Not applicable.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was signed by the patient’s parents for the genetic testing.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Y., Zhang, Y., Zhang, V.W. et al. Mutations in both SAMD9 and SLC19A2 genes caused complex phenotypes characterized by recurrent infection, dysphagia and profound deafness – a case report for dual diagnosis. BMC Pediatr 19, 364 (2019). https://doi.org/10.1186/s12887-019-1733-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1733-y