
Abstract
Background
Classical MPNs including ET and PMF have a chronic course and potential for leukaemic transformation. Timely diagnosis is obligatory to ensure appropriate management and positive outcomes. The aim of this study was to determine the mutational profile, clinical characteristics and outcome of ET and PMF patients in Pakistani population.
Methods
This was a prospective observational study conducted between 2012 and 2017 at NIBD. Patients were diagnosed and risk stratified according to international recommendations. Response to treatment was assessed by IWG criteria.
Results
Of the total 137 patients analysed, 75 were ET and 62 were PMF. JAK2 positivity was seen in 51 cases (37.2%), CALR in 41 cases (29.9%), while triple-negative in 17 (12.4%) cases. None of the patients in the present study were MPL positive. Overall survival for patients with ET and PMF was 92.5 and 86.0% respectively and leukaemia free survival was 100 and 91.6% respectively, at a median follow-up of 12 months. Leukaemic transformation occurred in 6.5% of MF patients; among them, JAK2 mutation was frequently found. Molecular mutations did not influence the OS in ET whereas in PMF, OS was shortest in the triple-negative PMF group as compared to the JAK2 and CALR positive patient groups.
Conclusion
This study shows a different spectrum of molecular mutations in ET and PMF patients in Pakistani population as compared to other Asian countries. Similarly, the risk of leukaemic transformation in ET and PMF is relatively lower in our population of patients. The factors responsible for these phenotypic and genotypic differences need to be analysed in large scale studies with longer follow-up of patients.
Similar content being viewed by others


Background
Primary Myelofibrosis and Essential thrombocythemia are classical Philadelphia-negative myeloproliferative neoplasms (MPNs), characterized by stem cell-derived clonal proliferation of one or more of myeloid lineage cells. The incidence of the classical MPNs reported worldwide is approximately 0.5–6/100,000 per year. It is considered a disease of the elderly with peak incidence occurring in the 5th to 6th decades of life [1, 2]. MPNs have the tendency to progress into myelofibrosis and transform into acute leukaemia after a certain period which may vary with each subtype of MPN [3].
The latest advancements in the molecular pathogenesis of classical MPN have revealed that each subtype of MPN carries a specific driver mutation including JAK2, CALR and MPL or somatic mutations in TET2, ASXL1, IDH, IKZF1, EZH2, DNMT3A, TP53, SF3B1, SRSF2, U2AF1 or other mutations [4]. The most recent revision of the classification of MPN published by the World Health Organization (WHO) has incorporated the presence of CALR and MPL mutations in the diagnostic criteria of PMF and ET based on the current evidences [5]. CALR mutations which are typically insertions or deletions and involve exon 9 have been reported in 60–90% of PMF and ET patients with unmutated JAK2 or MPL [6]. The most frequent subtypes of CALR are Type-1 (L367fs*46) and Type-2 (K385FS*47) [7]. It is generally believed that driver mutations are crucial for the MPN phenotype whereas the other mutations are associated with disease progression and leukaemic transformation [8].
The clinical presentation of ET is heterogeneous ranging from asymptomatic thrombocytosis to life threatening bleeding or thrombosis involving the major vessels of the body [9]. Patients who present with extreme thrombocytosis (> 1500 × 109/L) require vigilant monitoring because of the increased risk of haemorrhage due to acquired von Willebrand syndrome [10]. The risk of leukaemic transformation or progression into post-ET myelofibrosis increases with thrombosis, leucocytosis and increasing age [11]. On the other hand, typical clinical features of PMF include progressive anaemia, symptomatic splenomegaly, and various constitutional symptoms requiring treatment [12]. PMF is associated with a poor outcome and reduced life expectancy, with median survival durations ranging from 3.5 to 6 years, according to the previous studies [13]. Transformation into acute leukaemia occurs in approximately 20% of patients [14].
The diagnosis and management of MPNs in developing countries have always been challenging due to limited health resources. The molecular diagnostic facilities are limited to a few large tertiary care centres where access of patients from remote areas is difficult. Lack of awareness and delay in diagnosis results in suboptimal treatment, making the prognosis dismal in this part of the world.
In Pakistan, there is no well-defined cancer registry for MPN or other cancers, therefore data regarding the incidence, clinical presentation and outcome of patients suffering from different subtypes of MPN are scarce. Until 2012, molecular diagnostic facilities in our country were limited to PCR for BCR-ABL and JAK2 mutations. This is the first study from Pakistan which includes the molecular diagnosis of MPN based on cytogenetic analysis, PCR for JAK2, CALR and MPL mutations. The aim of this study was to determine the incidence, biological characteristics and clinical features in association with molecular mutations, and the overall survival and outcome of patients with ET and PMF, presenting to our tertiary care centre from all the major provinces of Pakistan.
Methods
Study design
The study was prospective observational and conducted at National Institute of Blood Diseases & Bone Marrow Transplantation between 2012 and 2017. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the ethics committee of NIBD and BMT (NIBD/RD-135/15–2012). Informed written consent was obtained from all patients before entering the data into the electronic database system.
Diagnosis
ET and PMF were diagnosed according to World Health Organization (WHO) classification of Myeloid and Lymphoid Malignancies 2008 [15]. Complete blood count (CBC), bone marrow biopsy and molecular and cytogenetic analyses were recorded for each patient. A symptom-assessment form (SAF) was given to all patients at baseline and subsequent visits to avoid subjectivity in the assessment of the degree of constitutional symptoms and the effects on the quality of life of patients. Measurements for liver and spleen size were also recorded.
Molecular and cytogenetic analysis
Cytogenetic analysis was performed using conventional G-banding techniques. The JAK2 mutation was assessed using a polymerase chain reaction (PCR)-based amplification system [16]. Sanger sequencing was performed to detect the MPL W515L/K and CALR exon 9 mutations. Exon 10 of MPL was amplified using the following primers: F, 5′-TTCTGTACATGAGCATT- TCATCA-3′ and R, 5′-GACAGGCTGTGTGTGTGTACCTCT-3′. Exon 9 of CALR was amplified using the following primers: F, 5′-GAGGAGTTTGGCAA CGAGAC-3′ and R, 5′-AACCAAAATCCACCCCAAAT-3′.
Risk stratification
Patients diagnosed with ET were categorized into high and low risk based on the presence or absence of thrombosis and age ≥ 60 years [17]. For patients with PMF, the DIPSS plus scoring system defined by the International Working Group (IWG) for MF was used to categorize patients into low, intermediate-1, intermediate-2 and high-risk groups [18].
Assessment of response and disease progression
The response to treatment in ET was assessed according to revised-response criteria proposed by IWG-MRT [19]. All patients received 300 mg of aspirin. Platelet pheresis was offered to patients with platelet counts ≥1500 × 109/L at baseline or those having thromboembolic manifestations regardless of platelet counts. Von Willebrand factor activity was checked in all patients with platelet counts of ≥1500 × 109/L, to rule out acquired von Willebrand disease. High risk patients received cytoreductive therapy with hydroxyurea along with aspirin. Pegylated interferon or oral busulfan was offered to those intolerant or resistant to first-line treatment.
Response assessment in PMF was based on revised-response criteria proposed by IWG-MRT and ELN, including normalization of blood counts and age-adjusted normocellularity of bone marrow, resolution of constitutional symptoms and hepatosplenomegaly after a treatment of at least ≥12 ± weeks [20]. For symptomatic splenomegaly, hydroxyurea and for anaemia, erythropoiesis stimulating agents in combination with synthetic androgens were used. JAK2 inhibitor was offered to few patients, when it received FDA approval in 2014. The presence of circulating blasts and changes in the grade of bone marrow fibrosis from baseline was considered as sign of disease progression into post-ET MF or acute leukaemia.
Statistical analysis
SPSS software (IBM SPSS Statistics, New York, USA, version 20.0) was used to calculate the frequency of qualitative variable i.e., gender and mean, median and standard deviation of quantitative variables such as age, haemoglobin, platelets and white blood cells. Continuous variables were analysed by using the Wilcoxon rank-sum test. Patient characteristics were compared using the Fisher’s exact test. Overall survival (OS) was defined as the time from diagnosis of ET or PMF to date of death (uncensored) or last contact (censored). Leukaemia-free survival (LFS) was calculated from the date of diagnosis to transformation into leukaemia. OS and LFS were plotted using Kaplan-Meier curves and compared by a log-rank test. P values< 0.05 were considered to indicate statistically significant differences.
Results
Frequencies of molecular and cytogenetic mutations
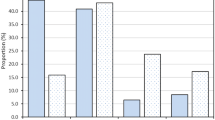
A total of 137 patients were analysed in this study, 75 patients were diagnosed with ET and 62 patients were diagnosed with PMF. JAK2 positivity was seen in 51 cases (37.2), CALR in 41 cases (29.9%), and triple-negative in 17 (12.4%) cases. Of the 75 patients with ET, 28 (37.3%) harboured the JAK2 mutation, and 22 (29.3%) harboured the CALR mutation. MPL mutation was not detected in any of the patients. Fourteen (18.7%) patients were triple-negative for all 3 mutations (Fig. 1). ET patients with CALR mutations accounted for 46.8% of patients who had non-mutated JAK2. Of the ET patients with CALR mutations, 13 (59.1%) had Type 1 mutation and 9 (40.9%) had Type 2 mutation.

Distribution of JAK2 V617F, MPL, and CALR mutations in patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF)
Of the 62 patients with PMF, 23 (37.1%) harboured the JAK2 mutation, 19 (30.6%) had CALR mutation and none of the patient harboured the MPL mutation. Three (4.8%) patients were negative for all 3 mutations (Fig. 1). PMF patients with CALR mutations accounted for 48.7% of the patients with non-mutated JAK2. Of those with mutated CALR, 52.6% had Type 1 CALR mutation while 47.7% had Type 2 CALR mutation. Homozygous CALR mutation was detected in one patient with the fibrotic phase of PMF, which was an exclusive finding, that has never been previously reported in MPN patients [21]. Six out of 7 patients with post-ET and post-PV MF harboured the JAK2 mutation.
Cytogenetic analysis revealed an abnormal karyotype in 10 (7.2%) patients. The most common karyotypic abnormality detected was del20q in 5% of patients followed by trisomy + 8 and + 13 in small number of PMF patients.
Clinico-haematologic features and genotype-phenotype correlation
Of patients with ET, 37 (49%) were male. According to 2013 ELN risk stratification, 52 (69.3%) were low risk patients and 23 (30.7%) were high risk patients. The median age of patients was 38 years (range: 19–56 years) and 71 years (range: 30–89 years) in the low and high-risk groups respectively. Splenomegaly was found in 35.7, 77.2 and 50% of JAK2 positive, CALR positive and triple-negative patients respectively. Table 1 summarizes the clinical and haematological characteristics of the study patients based on molecular mutations. Among the 3 mutational groups, JAK2 positive ET was associated with older age (58.5 ± 14.4 years) and large spleen size; CALR positive ET was associated with younger age (37 ± 10.4 years), higher platelet count (1191.9 ± 653.2 × 109/L) and low haemoglobin levels (11.6 ± 2.2 g/dl) and triple-negative ET was associated with higher WBC count (19.1 ± 36.9 × 109/L). Statistically significant differences were observed between the three groups for age (p-value: < 0.001) and spleen size (p-value: 0.007). Thromboembolic manifestations and constitutional symptoms were commonly observed in JAK2 positive ET.
Of patients with PMF, 34 (54.8%) were male. The median age of patients was 52 years (range: 20–81 years). The study characteristics of PMF patients are shown in Table 2. According to DIPSS plus risk stratification, 4 (6.5%) were low risk, 7 (11.3%) were intermediate-1 risk, 27% (43.5%) were intermediate-2 risk and 7 (11.3%) were high risk patients. Splenomegaly and circulating blasts were found in 75.8 and 6.5% of patients at baseline respectively. JAK2 positive PMF was associated with older age (53.0 ± 16.2 years) and intermediate-2 risk disease, whereas CALR positive PMF was also associated with intermediate-2 risk disease.
Triple-negative PMF was associated with the lowest haemoglobin (7.4 ± 1.2 g/dl) and platelet count (100.3 ± 62.0 × 109/L) and the highest WBC count (40.6 ± 66.9 × 109/L) among the 3 mutational groups.
Response to therapy and leukaemic transformation
Complete response to first-line treatment was achieved in 25 (48.1%) and 12 (52.5%) of low and high-risk ET patients respectively. Platelet-pheresis was required in 3 (5.8%) and 7 (30.4%) of low and high-risk patients respectively at initial diagnosis. Five (6.6%) patients were found refractory/resistant to first-line treatment and responded to second line treatment (pegylated-interferon). Progression into myelofibrosis occurred in 3 (4%) of patients but none of the patients transformed into acute leukaemia. Among patients with PMF, 13 (54.2%) patients showed a response to treatment with conventional agents. Twenty-four (63.2%) patients treated with JAK2 inhibitor showed a significant reduction in spleen size and improvement in constitutional symptoms. Leukaemic transformation was observed in 5 (8.1%) of patients.
Impact of molecular mutations on overall survival and prognosis
Overall survival for patients with ET and PMF was 92.5 and 86.0% respectively and leukaemia free survival for ET and MF was 100 and 91.6% respectively, at a median follow-up of 12 months (range:10–240 months) as shown in Fig. 2. None of the ET patients had leukaemic transformation while 8.1% of MF patients transformed and this transformation occurred more commonly in JAK2 positive patients (p value = 0.377). Figure 3a shows that OS in ET was not affected by molecular mutational status whereas in PMF, OS was shortest in triple-negative group of patients (p value = 0.053) as shown in Fig. 3b. Among the other clinical parameters, univariate analysis found that an intermediate-2 DIPSS score was associated with significantly shorter OS (p = 0.234) and LFS (p = 0.032) than the intermediate-1 or high-risk group. JAK2 mutation was associated with a higher risk of thromboembolic complications both in ET and PMF.

Overall Survival of the study participants

a Overall survival in essential thrombocythemia patients based on mutations. b Overall survival in primary myelofibrosis patients based on mutations
Discussion
Driver mutations such as JAK2, CALR and MPL contribute to the heterogeneity in the phenotypic behaviour and outcome in patients with different subtypes of MPN [22,23,24,25]. This study presents the clinical and molecular profiles of ET and PMF patients from different regions of Pakistan to understand the differences in clinical presentation between the Pakistani population and other countries.
Data concerning the molecular mutations in MPN from Pakistan are scarce. Most of the literature related to MPN from South East Asian countries is from China and South Korea. The frequency of JAK2 mutation reported in our study, is relatively lower than that reported in international studies; however the frequency of CALR and triple-negative MPN is consistent with those published in China and Korea. JAK2V617F was the first specific mutation identified in MPN pathogenesis, occurring with the highest frequency in polycythemia vera (81–99% of cases) followed by ET (41–72%) and MF (39–57%) and could be present as a heterozygous or homozygous mutation [26,27,28,29]. Bo Hyun Kim et al., reported the frequency of JAK2 (51.2, 54%), CALR (27.4, 22%) and triple-negative MPN (20.2, 20%), among 84 ET and 50 MF patients respectively, from Korea [30]. A similar study conducted by Li et al in 357 Chinese patients with PMF found that, 178 (50%) of patients carried JAK2V617F, 76 (21%) had a CALR mutation, 11 (3%) carried an MPL mutation, and 96 (27%) were triple-negative PMF [31]. Rumi et al., reported JAK2 (62%), CALR (24%), MPL (4%), and triple-negative ET (10%) among 745 European ET patients [32].
The incidence of CALR mutation in this study was in concordance with other studies from the Southeast Asian region but did not support the findings published in Western literature. Klampfl et al. initially reported a higher incidence of CALR mutations (67% in ET and 88% in PMF) in JAK2 and MPL negative patients [6]. All the mutations identified occurred in exon 9 of the CALR gene. The ratio of Type 1 versus Type 2 CALR mutation in our study corresponds to that found in PMF and ET patients in Asian and European countries except for China, where this ratio is reversed i.e. Type 2 mutation is more prevalent in the Chinese population [31]. The prognostic value of Type 1 and Type 2 mutations has been discussed in various studies. Tefferi et al. showed that patients who carry the Type 1 CAL-R mutation had significantly longer survival than the patients with all other driver mutations [33].
Unexpectedly, none of the patients with ET and PMF in our study harboured the MPL mutation. MPL mutations may occur in as many as 8% of ET and MF patients, although the actual frequency of MPL mutations in MPN patients has not been as extensively studied as the prevalence of JAK2 mutation [34]. Although very low frequency of MPL is reported in Korean population [30], the absence of MPL mutation in our population is a rare finding that needs confirmation in large scale studies.
The frequency of triple-negative MPN varies between 10 and 20% [35]. In our patients, triple-negativity was less commonly observed in PMF than ET. A European study reported 8.6% frequency of triple-negative PMF among 617 patients studied [36]. The ethnicity-based differences in the genetic profiles of the patients may be attributable to the incongruent findings observed in this study.
A small number of PMF patients in this study presented with cytogenetic abnormalities such as del20q and trisomy 8 at baseline. We did not find any statistically significant association of cytogenetic abnormalities with the molecular mutational profile of patients, and no clinical impact of these mutations could be observed on leukaemic transformation or overall survival of these patients. Approximately one third of patients with PMF present with cytogenetic abnormalities including del(20q), del(13q), trisomy 8 and 9, and abnormalities of chromosome 1 including duplication 1q. Patients with PMF that transform to acute leukaemia usually show complex karyotypes at transformation and a significantly decreased median survival [37, 38].
Overall, the clinical characteristics of our patients conformed to the results published in previous studies. In this study, JAK2 mutation was associated with older age, high-risk disease and increased incidence of thrombosis or haemorrhage compared to CALR positive and triple-negative ET and PMF. The association of JAK2 mutation with thromboembolism is well established in the literature. It is suggested that this mutation likely causes thrombosis through multiple mechanisms, including activation of platelets and granulocytes [39, 40]. More recently, the association of leucocytosis and JAK2 mutation with thrombotic events has been confirmed in a retrospective study of 108 patients with ET [41]. Increased rate of vascular complications in ET have been associated with two variables, age and previous thrombotic history [42].
CALR-mutant ET and PMF have relatively indolent clinical course compared with the respective JAK2-mutant disorders [32]. In this study, CALR mutation was associated with higher platelet count, lower leukocyte count and low-risk disease. These findings correlate with previously published study [43]. Three large cohort studies reported that an increased baseline leukocyte count was an independent risk factor for both thrombosis and inferior survival in ET [44]. This might explain the lower incidence of thrombotic events and better overall survival associated with CALR mutations in ET. A recent evaluation of 709 consecutive Mayo Clinic patients with PMF, confirmed that survival was significantly longer with Type 1 CALR, compared to all other driver mutations, which were otherwise similar in their prognosis [33].
In our study, triple-negative ET and PMF were associated with lower haemoglobin levels and higher WBC counts. Triple- negative ET had a less severe disease course. Triple-negative PMF had more constitutional symptoms, high-risk disease and increased incidence of thrombo-embolic events at baseline. The risk of leukaemic transformation in triple-negative PMF was higher than the JAK2 and CALR-mutated PMF in this study, leading to short OS in this group. These findings for triple-negative patients correlate with previously published studies from Asian and Western countries [45, 46]. Tefferi et al. have also highlighted the high-risk features of disease associated with triple-negative PMF [35].
Overall, the mutational status did not produce clinical impact on OS in ET, but in contrast, OS was found to be low in PMF patients who were triple-negative for all mutations as compared to JAK2 and CALR mutated patients.
Conclusion
This study shows a different spectrum of molecular mutations in ET and PMF patients in the Pakistani population compared to other Asian countries. Similarly, the risk of leukaemic transformation in ET and PMF is relatively lower in our population of patients. The factors responsible for these phenotypic and genotypic differences need to be analysed in large scale studies with longer follow up of patients.
The major limitations of this study include the relatively low numbers of patients in our cohort and lack of availability of next generation sequencing data for patients with triple-negative MPN.
Availability of data and materials
The datasets generated and analysed during the current study are not publicly available due to breach of confidentiality but are available from the corresponding author on reasonable request and after removing all the identifiable data.
Abbreviations
- CALR:
-
Calreticulin
- DIPSS:
-
Dynamic International Prognostic Scoring System
- ELN:
-
European Leukaemianet
- ET:
-
Essential thrombocythemia
- IWG - MRT:
-
International Working Group-Myeloproliferative Neoplasms Research and Treatment
- JAK2:
-
Janus Kinase 2
- LFS:
-
Leukaemia-free Survival
- MPL:
-
Thrombopoietin receptor gene
- MPNs:
-
Myeloproliferative Neoplasms
- OS:
-
Overall survival
- PMF:
-
Primary myelofibrosis
- SAF:
-
Symptom-assessment form
- WHO:
-
World Health Organization
References
Titmarsh GJ, Duncombe AS, McMullin MF, O'rorke M, Mesa R, De Vocht F, et al. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am J Hematol. 2014;89(6):581–7.
Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55(3):595–600.
Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507–13.
Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukaemia. 2010;24(6):1128–38.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukaemia. Blood. 2016;127(20):2391–405.
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–90.
Gold LI, Eggleton P, Sweetwyne MT, Van Duyn LB, Greives MR, Naylor S-M, et al. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010;24(3):665–83.
Barbui T, Thiele J, Gisslinger H, Finazzi G, Vannucchi A, Tefferi A. The 2016 revision of WHO classification of myeloproliferative neoplasms: clinical and molecular advances. Blood Rev. 2016;30(6):453–9.
Gisslinger H, editor. Update on diagnosis and management of essential thrombocythemia. Seminars in thrombosis and hemostasis; Copyright© 2006 by Thieme medical publishers, Inc., New York. 2006.
Besses C, Cervantes F, Pereira A, Florensa L, Sole F, Hernandez-Boluda J, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukaemia. 1999;13(2):150–4.
Passamonti F, Rumi E, Arcaini L, Boveri E, Elena C, Pietra D, et al. Prognostic factors for thrombosis, myelofibrosis, and leukaemia in essential thrombocythemia: a study of 605 patients. Haematologica. 2008;93(11):1645–51.
Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29(5):573–82.
Cervantes F, Passamonti F, Barosi G. Life expectancy and prognostic factors in the classic BCR/ABL-negative myeloproliferative disorders. Leukaemia. 2008;22(5):905–14.
Cervantes F. How I treat myelofibrosis. Blood. 2014;124(17):2635–42.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukaemia: rationale and important changes. Blood. 2009;114(5):937–51.
Chen Q, Lu P, Jones AV, Cross NC, Silver RT, Wang YL. Amplification refractory mutation system, a highly sensitive and simple polymerase chain reaction assay, for the detection of JAK2 V617F mutation in chronic myeloproliferative disorders. J Mol Diagn. 2007;9(2):272–6.
Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2012;87(3):284–93.
Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–8.
Barosi G, Mesa R, Finazzi G, Harrison C, Kiladjian J-J, Lengfelder E, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. 2013;121(23):4778–81.
Tefferi A, Cervantes F, Mesa R, Passamonti F, Verstovsek S, Vannucchi AM, et al. Revised response criteria for myelofibrosis: international working group-Myeloproliferative neoplasms research and treatment (IWG-MRT) and European LeukaemiaNet (ELN) consensus report. Blood. 2013;122(8):1395–8.
Rizvi Q, Zaidi U, Shahid S, Ahmed S, Shamsi T. Homozygous CALR mutation in primary Myelofibrosis and its effect on disease phenotype: a case report and review of the literature. Case Rep Hematol. 2019;2019:1–4.
Tefferi A, Vannucchi AM. Genetic Risk Assessment in Myeloproliferative Neoplasms. Mayo Clin Proc. 2017;92(8):1283-90. https://doi.org/10.1016/j.mayocp.2017.06.002.
Tefferi A, Lasho TL, Guglielmelli P, Finke CM, Rotunno G, Elala Y, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21–30.
Ortmann CA, Kent DG, Nangalia J, Silber Y, Wedge DC, Grinfeld J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372(7):601–12.
Vannucchi A, Lasho T, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukaemia. 2013;27(9):1861–9.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–97.
Kralovics R, Passamonti F, Buser AS, Teo S-S, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90.
James C, Ugo V, Le Couédic J-P, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–8.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–61.
Kim BH, Cho Y-U, Bae M-H, Jang S, Seo E-J, Chi H-S, et al. JAK2 V617F, MPL, and CALR mutations in Korean patients with essential thrombocythemia and primary myelofibrosis. J Korean Med Sci. 2015;30(7):882–8.
Li N, Yao Q-M, Gale RP, Li J-L, Li L-D, Zhao X-S, et al. Frequency and allele burden of CALR mutations in Chinese with essential thrombocythemia and primary myelofibrosis without JAK2V617F or MPL mutations. Leukaemia Res. 2015;39(5):510–4.
Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, Milosevic JD, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544–51.
Tefferi A, Nicolosi M, Mudireddy M, Szuber N, Finke CM, Lasho TL, et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1,095 patients. Am J Hematol. 2018;93(3):348–55.
Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–6.
Tefferi A, Lasho T, Finke C, Knudson R, Ketterling R, Hanson C, et al. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukaemia. 2014;28(7):1472–7.
Rumi E, Pietra D, Pascutto C, Guglielmelli P, Martínez-Trillos A, Casetti I, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124(7):1062–9.
Hussein K, Van Dyke DL, Tefferi A. Conventional cytogenetics in myelofibrosis: literature review and discussion. Eur J Haematol. 2009;82(5):329–38.
Tefferi A, Mesa RA, Schroeder G, Hanson CA, Li CY, Dewald GW. Cytogenetic findings and their clinical relevance in myelofibrosis with myeloid metaplasia. Br J Haematol. 2001;113(3):763–71.
Passamonti F, Rumi E, Pietra D, Della Porta MG, Boveri E, Pascutto C, et al. Relation between JAK2 (V617F) mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood. 2006;107(9):3676–82.
Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Villamor N, Colomer D, Cervantes F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica. 2006;91(2):169–75.
Kundranda MN, Maiti B, Iqbal N, Muslimani AA, Chaudhry A, Spiro TM, et al. The association of leukocytosis, thrombocytosis and JAK2V617F mutation with thrombotic events in myeloproliferative disorders (MPD’s). Blood. 2008;112(11):2803.
Finazzi G, Barbui T. Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukaemia. 2008;22(8):1494–502.
Rotunno G, Mannarelli C, Guglielmelli P, Pacilli A, Pancrazzi A, Pieri L, et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood. 2014;123(10):1552–5.
Barbui T, Carobbio A, Rambaldi A, Finazzi G. Perspectives on thrombosis in essential thrombocythemia and polycythemia vera: is leukocytosis a causative factor? Blood. 2009;114(4):759–63.
Tefferi A, Wassie EA, Guglielmelli P, Gangat N, Belachew AA, Lasho TL, et al. Type 1 versus type 2 calreticulin mutations in essential thrombocythemia: a collaborative study of 1027 patients. Am J Hematol. 2014;89(8):E121–E4.
Andrikovics H, Krahling T, Balassa K, Halm G, Bors A, Koszarska M, et al. Distinct clinical characteristics of myeloproliferative neoplasms with calreticulin mutations. Haematologica. 2014;99(7):1184–90.
Acknowledgements
All the patients, the healthcare professionals and laboratory staff are being acknowledged for their immense contribution into conducting of the present study. We also acknowledge the Springer Nature Author Services for their services rendered in betterment of the English of the manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
UZ- made substantial contributions to the conception of the work, drafted the work revised it critically for important intellectual content, approved the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; GS- made substantial contributions to the acquisition of data and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; MR- made substantial contributions to the acquisition of data and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; BK - made substantial contributions to the acquisition, analysis and interpretation of data and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; SMa made substantial contributions to the acquisition, analysis and interpretation of data and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; SNM - revised it critically for important intellectual content and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved;RZAK - revised it critically for important intellectual content and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; SMu - revised it critically for important intellectual content and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; MB - revised it critically for important intellectual content, approved the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved; TSS - revised it critically for important intellectual content, approved the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the ethics committee of NIBD and BMT (NIBD/RD-135/15–2013). Informed written consent was obtained from all patients before capturing the data in the electronic database system.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zaidi, U., Sufaida, G., Rashid, M. et al. A distinct molecular mutational profile and its clinical impact in essential thrombocythemia and primary myelofibrosis patients. BMC Cancer 20, 205 (2020). https://doi.org/10.1186/s12885-020-6700-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-6700-3