
Abstract
Background
There is growing evidence that pseudogenes may serve as prognostic biomarkers in several cancers. The present study was designed to develop and validate an accurate and robust pseudogene pairs-based signature for the prognosis of hepatocellular carcinoma (HCC).
Methods
RNA-sequencing data from 374 HCC patients with clinical follow-up information were obtained from the Cancer Genome Atlas (TCGA) database and used in this study. Survival-related pseudogene pairs were identified, and a signature model was constructed by Cox regression analysis (univariate and least absolute shrinkage and selection operator). All individuals were classified into high- and low-risk groups based on the optimal cutoff. Subgroups analysis of the novel signature was conducted and validated in an independent cohort. Pearson correlation analyses were carried out between the included pseudogenes and the protein-coding genes based on their expression levels. Enrichment analysis was performed to predict the possible role of the pseudogenes identified in the signature.
Results
A 19-pseudogene pair signature, which included 21 pseudogenes, was established. Patients in high-risk group demonstrated an increased the risk of adverse prognosis in the TCGA cohort and the external cohort (all P < 0.001). The novel pseudogene signature was independent of other conventional clinical variables used for survival prediction in HCC patients in the two cohorts revealed by the multivariate Cox regression analysis (all P < 0.001). Subgroup analysis further demonstrated the diagnostic value of the signature across different stages, grades, sexes, and age groups. The C-index of the prognostic signature was 0.761, which was not only higher than that of several previous risk models but was also much higher than that of a single age, sex, grade, and stage risk model. Furthermore, functional analysis revealed that the potential biological mechanisms mediated by these pseudogenes are primarily involved in cytokine receptor activity, T cell receptor signaling, chemokine signaling, NF-κB signaling, PD-L1 expression, and the PD-1 checkpoint pathway in cancer.
Conclusion
The novel proposed and validated pseudogene pair-based signature may serve as a valuable independent prognostic predictor for predicting survival of patients with HCC.
Similar content being viewed by others

Background
Hepatocellular carcinoma (HCC) is the most prevalent subtype of hepatic malignancies worldwide, accounting for 90% of primary liver cancers [1]. HCC is particularly prevalent in developing countries, particularly in East Asia and sub-Saharan Africa when compared with developed countries [2, 3]. Previous epidemiological studies have reported there to be approximately 250,000 new subjects and approximately 500,000 to 600,000 deaths due to HCC annually [1]. Despite the rapid advances in imaging techniques, surgical resection, and comprehensive therapy to treat HCC in recent years, the 5-year survival rate of HCC patients remains poor [4]. Therefore, it is necessary to uncover novel prognostic signatures that may identify groups of patients with a high risk of poor survival.
Pseudogenes are non-coding genes similar to their corresponding homologous protein-coding genes and long been considered ‘gene fossils’ or ‘junk genes’ because they do not encode functional proteins due to different kinds of mutations in the coding sequences [5]. In recent years, accumulating evidence has overwhelmingly revealed that individual pseudogenes involve in multiple human diseases including malignancy [6]. Multiple tumor-related pseudogenes have been confirmed as predictors for both diagnosis and prognosis. For example, the pseudogene DUXAP10 was found to be upregulated in several kinds of malignancies and could serve as a novel biomarker with high diagnostic and prognostic value for many cancers [7]. In HCC, high expression of the pseudogene ANXA2P2 has been found to be related to a worse prognosis. ANXA2P2 could be a novel predictive factor for evaluating the risk of recurrence or metastasis in HCC patients [8]. However, the molecular characteristics of pseudogene interactions and the prognostic value of pseudogenes in HCC have not been comprehensively explored.
Numerous studies have established mRNA expression profile-based signatures for outcome prediction in HCC patients [9,10,11,12,13,14]. However, these models have been failed to utilize clinically due to the diversity of data types, batch effects, and subsequent normalization of expression data, which poses a daunting obstacles for data processing given the possible biological heterogeneity among various data series and technical differences across different platforms [15]. Recently, a novel algorithm according to the relative orders of gene expression levels was established to remove the disadvantages of mRNA/miRNA expression normalization and scaling and has demonstrated robust results in previous studies [16, 17].
In this study, we identified 19 pseudogene-pairs based on univariate and LASSO regression analyses, and established a risk score model to predict the outcome of patients with HCC. Time-dependent receiver operating characteristic (ROC) curves were used to investigate the model’s performance in predicting the 1-, 3-, and 5-year overall survival (OS) of patients with HCC in two cohorts. Further, subgroup analysis was implemented to explore the prognostic performance of the signature in different stages, grades, sexes, and age groups. The C-index of the prognostic signature was compared with several established risk models. Pearson correlation analyses were done between the included pseudogenes and protein-coding genes based on their expression levels. Subsequently, we explored the biological functions and possible signaling pathways associated with the identified pseudogenes in the risk signature.
Methods
Data sources and pseudogene acquisition
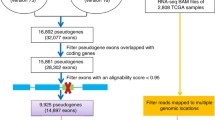
The most current 13,600 pseudogenes were searched from the HUGO Gene Nomenclature Committee (HGNC, https://www.genenames.org/download/statistics-and-files/). RNA-sequencing (RNA-seq) data from 374 HCC patients and 50 normal controls with corresponding clinical follow-up information (370 with complete follow-up clinical data) was screened out from the Cancer Genome Atlas (TCGA) database. Pseudogene expression levels were determined using the GENCODE project (http://www.gencodegenes.org) annotation by repurposing the probes in the RNA-seq expression profiles. Additionally, mRNA expression matrix and the clinical follow-up information for 240 patients with primary HCC (231 with complete follow-up information) and 202 normal controls were downloaded from the International Cancer Genome Consortium database (ICGC, https://dcc.icgc.org/, LIRI-JP) to validate the model externally. The probe IDs were changed to their gene symbols based on their annotation files without further standardization. For more than one probes corresponding to the same gene symbol, the probe average was calculated as the final expression value of gene. Patient ID numbers were matched with their gene expression profiles and follow-up data. The mRNA expression matrix of the shared pseudogenes was extracted from these two publicly available datasets.
Establishment of pseudogene pair-based prognostic signature
We first filtered out pseudogenes with imbalanced distribution or fairly little mutations [determined by median absolute deviation (MAD) < 0.5] across all samples in both cohorts [16]. Each pseudogene pair was analyzed by a pairwise comparison of pseudogene expression relative levels in a specific patient to obtain the score for per pseudogene pair. When the expression level of the first pseudogene more than the second pseudogene in a given pseudogene pair, the output value of the pseudogene pair was 1 and 0 for the different order, according to the proposed algorithm [16, 17]. Finally, 222 shared pseudogene pairs across two datasets were included. To explore the potential pseudogene pairs affecting the prognosis of HCC patients, univariate Cox regression analysis was used to identify the correlation between pseudogene pair expression and OS, with P < 0.05 being deemed statistically significant. Candidate factors were further screened by LASSO regression to yield the optimal informative but parsimonious model with 1000 iterations. Subsequently, a prognostic signature risk score was constructed according to the expression level of prognostic pseudogene pairs, weighted by the regression coefficient originated in the LASSO algorithm. Using the cutoff of the risk score generated by time-dependent ROC at 1 year for OS, all individuals were categorized into high- and low-risk groups.
Validation of the prognostic performance of the pseudogene pair model
Kaplan-Meier analysis along with a log-rank test was applied to compare the survival differences of the two risk groups. Time-dependent ROC curve analysis for OS was carried out to determine the predictive power of the model. Univariate Cox regression was performed to determine potential prognostic variables, and multivariate Cox analysis was perform to verify the effect of the risk score model on prognosis and other clinical factors. Hazard ratios (HRs) and their 95% confidence intervals (CIs) were estimated.
Comparison with other clinicopathological features and the novel prognostic model
To compare the effectiveness of the novel prognostic model with available clinicopathological factors and the recently built prognostic models, a comparison was implemented using the rcorrp.cens package in Hmisc in R and evaluated by C-index with 1000 bootstrap resamples.
Identification and enrichment analysis of pseudogene-related protein-coding genes
The Pearson correlation coefficients (|Pearson correlation coefficient| > 0.6 and P-value < 0.001) between the final identified pseudogenes and protein-coding genes were measured to detect their co-expression associations [18]. Gene Ontology (GO) functional enrichment analysis as well as Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were also conducted utilizing the clusterProfiler package to investigate the biological function and pathways involving numerous genes [19].
Statistical analysis
Survival curves were generated using the Kaplan–Meier method along with the log-rank test. Receiver operating characteristic (ROC) curves were generated using the R package “survivalROC”. The area under the curve (AUC) value obtained from the ROC curve was used to explore the diagnostic effectiveness of signature risk score in discriminating HCC tissues from normal tissues in two cohorts. Multivariate analyses were carried out utilizing the Cox proportional hazards regression model. A P-value less than 0.05 was considered significant.
Results
Establishing the pseudogene pair-based signature
The follow-up clinical information of patients in the two cohorts were shown in Table 1. A total of 222 pseudogene pairs were identified from 36 shared pseudogenes in the TCGA cohort after filtering by MAD > 0.5 as mentioned above. Univariate Cox regression analysis was carried out for the 222 pseudogene pairs to reveal 38 pseudogene pairs presenting significant prognostic potential (P < 0.05). Next, we performed LASSO Cox regression algorithm to reduce the number of pseudogene pairs in the risk model. After 1000 iterations, 19 pseudogene pairs were obtained and used to build a prognostic risk signature (Fig. 1). The risk signature consisted of 21 unique pseudogenes (Table 2).

Predictor selection by LASSO algorithm. a: Parameter filter by LASSO regress algorithm used five-fold cross-validation by through minimum criteria; b: Optimal feature selection based on LASSO coefficient profile plot of 19 pseudogene pairs
Association between signature risk score and clinical characteristics
To confirm the clinical value of the pseudogene pair-based signature risk score, the Chi-square test was applied to assess the association between the risk score and available clinical parameters. In the TCGA cohort, a higher risk score was revealed to be associated notably with grade (III + IV vs grade I + II, P = 0.0021; Fig. 2a) and stage (III + IV vs I + II, P = 0.00043; Fig. 2b). However, no significant difference was found in age (P = 0.0021; Fig. 2c) and gender (P = 0.0021; Fig. 2d).

Association between the pseudogene pair-based signature risk score and clinical parameters in the TCGA cohort
Validation and assessment of the established signature
Next, the risk score of the novel signature for per patient were calculated in the TCGA cohort. The optimal cutoff score for classifying patients into high- or low-risk groups was determined as 0.509 employing time-dependent ROC curve analysis at 1 year for OS predication (Fig. 3). High-risk patients exhibited a worse prognosis than low-risk patients, as revealed by Kaplan-Meier and log-rank tests (HR: 5.12, 95% CI: 3.54.7.39, P < 0.001, Fig. 4a). Patients in high-risk group also had worse outcomes than low-risk patients in the ICGC cohort (HR = 3.2, 95%CI: 1.61–6.37, P < 0.001, Fig. 4b) using the same cutoff point as in the TCGA dataset.

Time-dependent ROC curve analysis of the risk score. A cutoff point of risk score was identified as 0.509 to divide patients into two distinct groups in the TCGA cohort

Kaplan-Meier survival curves for patients with HCC in two distinct groups. Survival cures in the TCGA cohort (a), ICGC dataset (b), and subgroup analysis with respect to age (c, d), gender (e, f), histological grade (g, h), American Joint Committee on Cancer stage (i, j), family history (k, l), and vascular invasion (m, n)
To evaluate the prognostic performance of the signature in different subgroups, we investigated the relationship between clinical pathological factors and the prognostic signature using Kaplan-Meier and log-rank tests. As shown in Fig. 4c-n, the Kaplan–Meier curves illustrated that the signature was a robust prognostic predictor for patients with HCC grouped by sex (male or female), age (< 60 years or ≥ 60 years), family history (Yes or No),grade (grade I-II or grade III-IV), vascular invasion (Yes or No), and stage (stage I-II or stage III-IV). Multivariate Cox regression analyses were used to screen out the independent predictor in two cohorts. After adjusting for other clinical and pathological variables, the prognostic signature risk score was still an independent prognostic variable for OS in the TCGA cohort (HR = 3.416, 95%CI: 2.551–4.576; P < 0.001) and was validated in the ICGC cohort (HR = 1.902, 95%CI: 1.201–3.014, P = 0.006, Table 3).
Furthermore, the AUC values of the prognostic model for the 1-, 3-, and 5-year survival rates prediction in the TCGA cohort were 0.78, 0.81, and 0.74, respectively, (Fig. 5a). This revealed the predictive performance of the prognostic signature to be quite promising. The AUC values for OS in the ICGC cohort at 1 year and 3 years were 0.71 and 0.67, respectively (Fig. 5b). These findings confirmed that the novel model accurately predicted the prognosis of patients with HCC.

The ROC curve for 1-, 3- and 5-year overall survival prediction using the pseudogene pair-based prognostic. a TCGA cohort; b ICGC cohort
To explore the diagnostic value of pseudogene pair-based signature, we generated a ROC curve using the risk score from 374 HCC patients and 50 healthy controls. The AUC was 0.839 (95%CI = 0.801–0.875; Fig. 6a), which was further confirmed in the ICGC cohort with an AUC of 0.871 (95%CI = 0.836–0.901; Fig. 6b). Subgroup analysis demonstrated the diagnostic value of signature risk score in early stage of HCC were robust with AUC value of 0.778 (95%CI = 0.720–0.829; Fig. 6c) for stage I disease in the TCGA cohort. The diagnostic power was confirmed in the ICGC cohort with an AUC of 0.872 (95%CI = 0.825–0.910; Fig. 6d) for stage I disease. These demonstrated that the pseudogene pair-based signature risk score had an excellent diagnostic value in discriminating HCC from normal samples.

Diagnosis value of pseudogene pair-based signature risk score in HCC and normal controls. ROC in normal tissues and HCC samples in the TCGA cohort (a) and ICGC cohort (b). ROC for stage I samples and normal tissues in the TCGA cohort (c) and ICGC cohort (d)
Comparison with previous existed prognostic signatures
We compared our novel model with previous established prognostic signatures and confirmed the predictive performance and precision of the signature. Most importantly, the novel signature yielded a C-index of 0.761, which was higher than that of risk models based on single variable, which included age, grade, sex, stage as well as the merged models (all P < 0.05, Fig. 7). Furthermore, we also compared our model with recent existing signatures used to predict HCC survival. The C-index of our prognostic signature was larger than that of previous existed models (all P < 0.05). In addition, the C-index of the signature combined with other variables was 0.774. Thus, a combination of our prognostic signature and other variables should provide a more accurate prediction. Therefore, the novel prognostic signature was robust in predicting the prognosis of HCC patients.

Comparison of C-index among the novel model, previously established prognostic signatures, and clinical features (age, sex, stage, grade, and their combination)
Functional analysis of co-expression genes
To further example the potential biological roles of the 21 unique pseudogenes identified, the protein-coding genes positively or negatively correlated with them (|Pearson correlation coefficient| > 0.6 and P-value < 0.001) were considered pseudogene-related protein-coding genes. A total of 842 genes were considered eligible for pathway enrichment. We conducted GO and KEGG enrichment analyses to uncover specific functional categories of the co-expressed genes. They were primarily involved in cytokine receptor activity, cytokine binding, chemokine receptor activity, C-C chemokine receptor activity, and chemokine binding (Table 4). KEGG pathway enrichment revealed that these genes were primarily involved in T cell receptor signaling, chemokine signaling, B cell receptor signaling, PD-L1 expression, NF-κB signaling, and the PD-1 checkpoint pathway in cancer (Table 4).
Discussion
HCC remains a major and growing global public health challenge. However, the molecular pathogenesis of HCC is not fully understood. Given the extensive heterogeneity of HCC, there is a need for more accurate individualized prognostic signatures. Recently, increasing evidence has demonstrated that abnormal expression of pseudogenes is involved in multiple diseases, including malignancy [6]. For example, in HCC, upregulation of the pseudogene RP11-564D11.3 has been found to be associated with adverse survival [20]. Numerous researches have built gene expression profile-based signatures for survival prediction in patients with HCC [9,10,11,12,13,14]. However, previous reports aiming to build a prognostic model have focused on mRNAs, lncRNAs, and miRNAs, neglecting pseudogenes as potential biomarkers in HCC. Therefore, the development of a robust pseudogene pair signature contributes to clinical decision-making for individualized treatment of HCC patients.
In this study, we established a novel 19-pseudogene pair signature that could successfully classify patients into two groups with different OS. We found that patients in high-risk group had a worse survival rate than patients in the low-risk group in both cohorts. Subgroup analysis by age, family history, sex, grade, vascular invasion, and stage yielded the same conclusion. We found the signature to be a stable prognostic predictor for patients with HCC. Multivariate analyses demonstrated that the risk score may be a clinically independent prognostic predictor for HCC. The AUC values of the prognostic model for OS prediction also present excellent predictive performance in both cohorts. The signature was reproducible and robust in the independent validation cohort, demonstrating its value and effectiveness. These conclusions confirmed that the novel model could offer an accurate survival prediction for patients with HCC. Moreover, the C-index of our signature was larger than that of established signatures. We employed a more comprehensive and novel approach to develop a robust prognostic signature for HCC and successfully validated it in the ICGC cohort. Therefore, this novel prognostic model is accurate, robust, and interpretable.
Although numerous prognostic models have been established for the prediction of HCC survival [9,10,11,12,13,14, 21, 22], these prognostic models have seldom been widely utilized clinically due to their need for proper data standardization across various expression profiles for further analysis [16, 17]. In this study, based on the relative orders of the mRNA expression, the signature was generated only by weight-pairwise comparison within a given sample without requiring for data normalization and can remove the batch effects between different platforms. Furthermore, the cutoff value derived from the risk score formula used in this study could be employed across multiple datasets, showing a great advantage when compared with previous models, and may be easily translated into clinical application. This novel algorithm has been validated to be accurate and robust in previous cancer-related reports [16, 17, 23, 24].
The identified pseudogene-related protein-coding genes were primarily involved in cytokine and chemokine receptor activity, and cancer-related pathways, such as T cell receptor signaling, NF-κB signaling, PD-L1 expression, and PD-1 checkpoint pathway in cancer. It has also been reported that IL-8 and IL-6, important chemokines, are involved in tumor angiogenesis, growth and metastasis, and can therefore act as vital chemokines for blood vessel formation in HCC [25,26,27]. Previous studies have confirmed that T cells gather in the blood of HCC patients, and tumor necrosis factor (TNF), which is involve in the T cell receptor signaling pathway, was downregulated in sorafenib-treated HCC patients, demonstrating that the T cell receptor signaling pathway may also be involved in HCC [28, 29]. A previous study has provided evidence for the inhibitory effect of PPARα on HCC via the NF-κB signaling pathway [30]. IGFBP2 can serve as a new therapeutic target that activates the NF-κB-ZEB1 signaling axis and contributes to HCC tumorigenesis [31]. Expression of immune checkpoint molecules, such as PD-1/PD-L1, has been confirmed in HCC [32]. Furthermore, in September 2017, the FDA has granted accelerated approval to PD-1 checkpoint inhibitors for the treatment of HCC patients [33]. Therefore, the novel established pseudogene pair signature could be associated with HCC-related biological pathways and the functional dysregulation could be well associated with the survival of HCC patients.
The signature based on the relative expression ordering exhibited no difficulty in clinical transformation and application. For future study, if the expression matrix of 21 pseudogenes was obtained, a 19-pseudogene pair signature was therefore constructed. Furthermore, the signature only involves pairwise comparison within a given sample without the requiring for data normalization and batch effects from different laboratories and platforms. Based on the cutoff value derived from the risk score formula, patients can be grouped into different groups, and their prognosis can be predicted. Thus, the prognostic model can act as an individualized, single-sample prediction of outcome of HCC and can be easily translated to clinical application.
This is the first research to establish a pseudogene pair-based prognostic signature in HCC. However, this study also has limitations. First, prospective research is suggested to validate the prognostic function of the pseudogene pair signature. Moreover, to better understand the functional role of these pseudogene pairs in HCC, experimental studies investigating these pseudogenes should be carried out.
Conclusion
We developed and validated an accurate and novel robust pseudogene pair signature capable of accurately predicting the prognosis of HCC patients, with higher risk scores demonstrating adverse prognosis. The signature is reproducible and robust in an independent external cohort and outperforms other established signatures, demonstrating its value and effectiveness. Additionally, this signature could act as an encouraging independent prognostic predictor for HCC.
Availability of data and materials
All the data used in the study are obtained from the TCGA database (https://portal.gdc.cancer.gov/) and ICGC database (https://dcc.icgc.org/), which are opening and available to all.
Abbreviations
- TCGA:
-
The Cancer Genome Atlas
- HCC:
-
Hepatocellular carcinoma
- LASSO:
-
Least absolute shrinkage and selection operator
- ROC:
-
Receiver operating characteristic
- OS:
-
Overall survival
- GO:
-
Gene Ontology
- HR:
-
Hazard ratios
- C-index:
-
Concordance index
- CI:
-
Confidence interval
- AUC:
-
Area under the curve
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet (London, England). 2012;379(9822):1245–55.
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–76.
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108.
Villanueva A, Hernandez-Gea V, Llovet JM. Medical therapies for hepatocellular carcinoma: a critical view of the evidence. Nat Rev Gastroenterol Hepatol. 2013;10(1):34–42.
Milligan MJ, Harvey E, Yu A, Morgan AL, Smith DL, Zhang E, Berengut J, Sivananthan J, Subramaniam R, Skoric A, et al. Global Intersection of Long Non-Coding RNAs with Processed and Unprocessed Pseudogenes in the Human Genome. Front Genet. 2016;7:26..
Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA (New York, NY). 2011;17(5):792–8.
Yue C, Ren Y, Ge H, Yan L, Xu Y, Wang G, Wu J. Pseudogene DUXAP10 can be used as a diagnostic and prognostic biomarker in human cancers. J Cell Physiol. 2019;234(12):23685–94.
Wang QS, Shi LL, Sun F, Zhang YF, Chen RW, Yang SL, Hu JL. High expression of ANXA2 Pseudogene ANXA2P2 promotes an aggressive phenotype in hepatocellular carcinoma. Dis Markers. 2019;2019:9267046.
Qiao GJ, Chen L, Wu JC, Li ZR. Identification of an eight-gene signature for survival prediction for patients with hepatocellular carcinoma based on integrated bioinformatics analysis. Peer J. 2019;7:e6548.
Shi YM, Li YY, Lin JY, Zheng L, Zhu YM, Huang J. The discovery of a novel eight-mRNA-lncRNA signature predicting survival of hepatocellular carcinoma patients. J Cell Biochem. 2019;120:7539–50.
Wang Y, Ruan Z, Yu S, Tian T, Liang X, Jing L, Li W, Wang X, Xiang L, Claret FX, et al. A four-methylated mRNA signature-based risk score system predicts survival in patients with hepatocellular carcinoma. Aging. 2019;11(1):160–73.
Wang Z, Teng D, Li Y, Hu Z, Liu L, Zheng H. A six-gene-based prognostic signature for hepatocellular carcinoma overall survival prediction. Life Sci. 2018;203:83–91.
Yan Y, Lu Y, Mao K, Zhang M, Liu H, Zhou Q, Lin J, Zhang J, Wang J, Xiao Z. Identification and validation of a prognostic four-genes signature for hepatocellular carcinoma: integrated ceRNA network analysis. Hepatol Int. 2019;13(5):618–30.
Zhang BH, Yang J, Jiang L, Lyu T, Kong LX, Tan YF, Li B, Zhu YF, Xi AY, Xu X, et al. Development and validation of a 14-gene signature for prognosis prediction in hepatocellular carcinoma. Genomics. 2020;112(4):2763–71.
Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–9.
Heinäniemi M, Nykter M, Kramer R, Wienecke-Baldacchino A, Sinkkonen L, Zhou JX, Kreisberg R, Kauffman SA, Huang S, Shmulevich I. Gene-pair expression signatures reveal lineage control. Nat Methods. 2013;10(6):577–83.
Li B, Cui Y, Diehn M, Li R. Development and validation of an individualized immune prognostic signature in early-stage nonsquamous non-small cell lung Cancer. JAMA Oncol. 2017;3(11):1529–37.
Bhardwaj N, Lu H. Correlation between gene expression profiles and protein-protein interactions within and across genomes. Bioinformatics (Oxford, England). 2005;21(11):2730–8.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–7.
Song H, Yang J, Zhang Y, Zhou J, Li Y, Hao X. Integrated analysis of pseudogene RP11-564D11.3 expression and its potential roles in hepatocellular carcinoma. Epigenomics. 2019;11(3):267–80.
Li N, Zhao L, Guo C, Liu C, Liu Y. Identification of a novel DNA repair-related prognostic signature predicting survival of patients with hepatocellular carcinoma. Cancer Manag Res. 2019;11:7473–84.
Liu GM, Zeng HD, Zhang CY, Xu JW. Identification of a six-gene signature predicting overall survival for hepatocellular carcinoma. Cancer Cell Int. 2019;19:138.
Kang CM, Jia XH, Liu HS. Development and validation of a RNA binding protein gene pair-associated prognostic signature for prediction of overall survival in hepatocellular carcinoma. Biomed Eng Online. 2020;19(1):68.
Zhao EF, Zhou CL, Chen SM: A signature of 14 immune-related gene pairs predicts overall survival in gastric cancer. Clin Transl Oncol 2020, https://doi.org/https://doi.org/10.1007/s12094-020-02414-7.
Zhu B, Lin N, Zhang M, Zhu Y, Cheng H, Chen S, Ling Y, Pan W, Xu R. Activated hepatic stellate cells promote angiogenesis via interleukin-8 in hepatocellular carcinoma. J Transl Med. 2015;13:365.
Ferguson-Smith AC, Chen YF, Newman MS, May LT, Sehgal PB, Ruddle FH. Regional localization of the interferon-beta 2/B-cell stimulatory factor 2/hepatocyte stimulating factor gene to human chromosome 7p15-p21. Genomics. 1988;2(3):203–8.
Tang KH, Ma S, Lee TK, Chan YP, Kwan PS, Tong CM, Ng IO, Man K, To KF, Lai PB, et al. CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology. 2012;55(3):807–20.
Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65(6):2457–64.
Liu Y, Wang P, Li S, Yin L, Shen H, Liu R. Interaction of key pathways in sorafenib-treated hepatocellular carcinoma based on a PCR-array. Int J Clin Exp Pathol. 2015;8(3):3027–35.
Zhang N, Chu ES, Zhang J, Li X, Liang Q, Chen J, Chen M, Teoh N, Farrell G, Sung JJ, et al. Peroxisome proliferator activated receptor alpha inhibits hepatocarcinogenesis through mediating NF-κB signaling pathway. Oncotarget. 2014;5(18):8330–40.
Guo Q, Yu DY, Yang ZF, Liu DY, Cao HQ, Liao XW. IGFBP2 upregulates ZEB1 expression and promotes hepatocellular carcinoma progression through NF-κB signaling pathway. Dig Liver Dis. 2020;52(5):573–81.
Hato T, Zhu AX, Duda DG. Rationally combining anti-VEGF therapy with checkpoint inhibitors in hepatocellular carcinoma. Immunotherapy. 2016;8(3):299–313.
Sun C, Lan P, Han Q, Huang M, Zhang Z, Xu G, Song J, Wang J, Wei H, Zhang J, et al. Oncofetal gene SALL4 reactivation by hepatitis B virus counteracts miR-200c in PD-L1-induced T cell exhaustion. Nat Commun. 2018;9(1):1241.
Acknowledgements
The author thank Editage (www.editage.cn) for English language editing.
Funding
None.
Author information
Authors and Affiliations
Contributions
Study concept and design: YJD and YG; data analysis/interpretation: YJD; manuscript drafting: YJD; manuscript revision: YJD and YG; approval of final version of submitted manuscript: YJD and YG;
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, Y., Gao, Y. Development and validation of a novel pseudogene pair-based prognostic signature for prediction of overall survival in patients with hepatocellular carcinoma. BMC Cancer 20, 887 (2020). https://doi.org/10.1186/s12885-020-07391-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-07391-2