
Abstract
Background
Non-small cell lung cancer is the most common cause of cancer death worldwide, highlighting the need for novel therapeutic concepts. In particular, there is still a lack of treatment strategies for the group of elderly and frail patients, who are frequently not capable of receiving standard therapy regimens. Despite comprising the majority of lung cancer patients, this group is underrepresented in clinical trials. This applies also to elderly and frail patients suffering from unresectable stage III NSCLC, who are unfit for chemotherapy, and, therefore, cannot receive the standard therapy comprising of radiochemotherapy and the recently approved subsequent durvalumab consolidation therapy. These patients often receive radiotherapy only, which raises the concern of undertreatment. The TRADE-hypo trial aims at optimizing treatment of this patient group by combining radiotherapy with concomitant durvalumab administration, thereby employing the immune-promoting effects of radiotherapy, and determining safety, feasibility, and efficacy of this treatment.
Methods/ design
In this prospective phase II clinical trial, durvalumab therapy will be combined with either conventionally fractionated (CON-group) or hypofractionated (HYPO-group) thoracic radiotherapy. A safety stop-and-go lead-in phase will assess safety of hypofractionated radiotherapy with respect to severe pneumonitis in small patient cohorts before opening full enrollment. Tumor tissue, blood and stool samples will be collected before and during the study period to investigate the immunological mechanisms responsible for checkpoint inhibitor efficacy and immune-promoting effects of radiotherapy.
Discussion
Preclinical data suggests that irradiation-induced immunogenicity can be further increased if applied in a hypofractionated setting, potentially boosting the expected synergistic effect with immune checkpoint inhibition in restoring the immune anti-tumor response. If proven safe and efficient, a hypofractionated radiation schedule can provide a considerably more practicable option for the patient. Taking into consideration the intend to develop a combination treatment strategy that can be made available to patients soon after proving to be efficient and the potentially elevated toxicity of a hypofractionated radiotherapy approach, this trial was designed as a two-trials-in-one design. An accompanying translational research program is planned striving to gain insights into the tumor-host biology and to identify suitable biomarkers to predict therapy response.
Trial registration
Clinicaltrials.gov, NCT04351256. Registered 17 April 2020,
Eudra-CT, 2019–002192-33. Registered 24 October 2019,
Similar content being viewed by others


Background
Lung cancer is the most common cause of cancer death worldwide, with non-small cell lung cancer (NSCLC) representing 80–90% of cases [1, 2]. Improving therapeutic strategies is thus of imminent importance, especially considering elderly and frail patients. With a median age of about 70 years at diagnosis lung cancer clearly is a disease of the elderly, yet this group is underrepresented in clinical trials, and these patients are frequently not capable of receiving standard treatment protocols due to age- and tobacco-associated comorbidities [3,4,5].
In recent years, the advent of immunotherapy has paved the way for novel therapeutic concepts, including the combination of radiotherapy with immune checkpoint inhibition (e.g. Programmed cell death/ -ligand 1; PD-1/ PD-L1). This approach is of particular interest as it utilizes synergistic effects: While immune checkpoint inhibitors can restore the patients’ antitumor immunity through T cell activation, radiotherapy may further boost immune-mediated anti-cancer mechanisms by exposing tumor-associated antigens and by attracting both immunocompetent antigen-presenting cells and tumoricidal effector cells [6, 7]. Indeed, for patients with unresectable stage III NSCLC, the PACIFIC trial has revealed a profound clinical benefit treatment with the anti-PD-L1 monoclonal antibody durvalumab after chemoradiotherapy with remarkably low toxicities [8, 9]. As a result, sequential treatment with durvalumab after chemoradiotherapy has become the new standard treatment for locally advanced, unresectable NSCLC. However, about 20% of patients do not receive chemotherapeutic agents, presumably due to significantly higher rates of age- and comorbidity-related adverse events (AE) under chemoradiotherapy [5, 10]. Thus, elderly and frail patients often receive radiotherapy alone, raising the serious concern of undertreatment and the need for new therapeutic concepts [4, 5].
Considering the immune-promoting effects of radiotherapy, a combination with durvalumab therapy may improve response rates in these potentially undertreated patients. Moreover, if applied early, concomitant local radiotherapy with systemic immunotherapy may particularly increase control of distant micrometastases. Preclinical data suggest that irradiation-induced immunogenicity can even be further increased if applied in a hypofractionated setting with single doses ≥3 Gray (Gy), in line with a radiation dose-dependent abscopal effect [11,12,13,14]. While a hypofractionated radiation schedule is also considerably shorter and more convenient for the patient, safety of concurrent immunoradiotherapy is a concern, as both therapy modalities may cause severe pneumonitis.
In this prospective phase II clinical trial, we therefore aim to determine feasibility and treatment efficacy of durvalumab treatment combined with thoracic radiotherapy (TRT) in previously untreated NSCLC stage III patients unable to receive radiochemotherapy. Striving to develop a combination treatment strategy that, if proven safe and efficient, can be quickly made available to patients, a two-trials-in-one design was chosen that combines durvalumab with either conventionally fractionated (CON-group) or hypofractionated thoracic radiotherapy (HYPO-group). This study not only aims to increase the efficacy of radiotherapy by utilizing the immune-sensitizing effects elicited by PD-L1 inhibition, but will also provide biomaterials that will be analyzed with respect to immunological mechanisms responsible for checkpoint inhibitor efficacy and immune-promoting effects of radiotherapy as well as potential biomarkers.
Methods/design
Study design
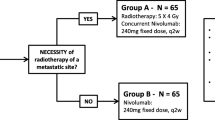
The TRADE-hypo trial is a prospective, randomized, open-label, multicenter, phase II trial with a safety stop-and-go lead-in phase (Fig. 1). During the lead-in phase, patients in the HYPO-group, who will receive durvalumab in combination with hypofractionated thoracic radiotherapy, will be closely evaluated with regard to toxicity (defined as pneumonitis ≥ grade 3 within 8 weeks after radiotherapy) in small cohorts (n = 6) before proceeding with full enrollment into this arm (Fig. 2).

Study design of the TRADE-hypo trial. Patients will be enrolled according to eligibility criteria and treated with either a hypofractionated TRT regimen (HYPO-group) or conventionally fractionated TRT (CON-group) in combination with durvalumab. For the HYPO-group, a safety stop-and-go phase with a 6 + 6 design precedes full enrollment. Whenever this arm is open for recruitment, patients will be allocated to this arm until the cohort is closed; whenever HYPO-arm is closed for Stop/ Go decision evaluation based on the toxicity assessment of this regimen 8 weeks after the end of TRT, patients are allocated to the CON-arm. When the study proceeds to expansion phase, patients will be allocated to treatment arms by randomization using “biased coin” algorithm. An efficacy interim analysis will be performed after 18 patients have been enrolled in each arm

Cohort design of the safety stop-and-go lead-in phase (HYPO-group). The safety lead-in phase follows a 6 + 6 design in order to carefully evaluate the toxicity of the treatment in the HYPO-group with respect of the occurrence of a grade 3/4 pneumonitis (“event”) within 8 weeks after the end of TRT. Two events in the first six patients, two events in the first 12, or two events in the first 18 patients will result in termination of the HYPO-group (“Stop”). If no event is observed within the first two safety cohorts, i.e. the first 12 patients, the HYPO-arm will be opened for full enrollment with close toxicity assessment with respect to pneumonitis grade 3/4, and terminated as soon as two events are reported within the subsequent six patients (“Go*”). Full enrollment in the HYPO-arm will only take place if the criteria for the non-toxicity scenario are met, i.e. ≤ 1 event in n = 18 patients (“Go”)
Study setting
The TRADE-hypo trial will recruit patients from 17 participating centers across Germany over a period of 20 months. Start of recruitment was planned for April 2020, but was delayed to May 2020 due to the Covid-19 pandemic. A full list of sites can be obtained at clinicaltrials.gov (NCT04351256).
Study objectives
The primary objective of this study is to evaluate safety and tolerability of conventionally fractionated (CON-group) and hypofractionated (HYPO-group) TRT in combination with durvalumab in patients with unresectable stage III NSCLC unfit for chemotherapy. Moreover, efficacies of the two modes of radiotherapy will be evaluated with respect to response rates. Further parameters will be determined in order to assess efficacy, safety, and quality of life (QoL) in both treatment arms by recording incidence and severity of adverse events (AEs) as well as specific laboratory abnormalities.
Exploratory endpoints include assessment of vulnerability and analyses of tumor tissue, blood, and stool samples that are collected during the clinical trial with respect to treatment-induced changes and immune-related biomarkers.
Characteristics of participants
A total of 88 patients will be included into this study. Patients potentially eligible for trial inclusion will be approached and asked to participate as they present in the clinic. Before a patient’s participation in the clinical study, the investigator must obtain written informed consent.
Each participant must be eligible regarding all inclusion and exclusion criteria set for this trial (Table 1). Key inclusion criteria include diagnosis of unresectable stage III NSCLC and non-feasibility of sequential chemoradiotherapy due to increased (oxygen-independent) vulnerability as reflected by fulfilling at least one of the following criteria: (i) Performance status 2 (Eastern Cooperative Oncology Group [ECOG] scale), (ii) ECOG 1 and Charlson comorbidity index (CCI) ≥ 1, or (iii) age ≥ 70. Moreover, participants must have at least one measurable site of disease as defined by RECIST 1.1, as well as adequate bone marrow, hepatic and renal function. Patients having received prior immunotherapy, other investigational agents or thoracic radiotherapy within the past 5 years will be excluded from the study. Additionally, participants must not have an active or recent history of a known or suspected autoimmune disease or any other medical condition conflicting with the study interventions, and not have used immunosuppressive medication. For a full list of the inclusion and exclusion criteria see Table 1.
Treatment
Durvalumab
Patients will be enrolled based on the in−/ exclusion criteria. Two treatment groups will be evaluated: One group will receive durvalumab immunotherapy combined with conventionally fractionated TRT (CON-group) and the other one with hypofractionated TRT (HYPO-group). In both groups, durvalumab will be administered intravenously at a fixed dose of 1500 mg on day 1 and every 4 weeks thereafter for a maximum of 12 months (maximum 13 doses, last dose at week 49) until confirmed disease progression, inacceptable toxicity, withdrawal of consent or end of the study (Fig. 1 and Table 2).
Radiotherapy
All patients are subjected to preparation of individual positioning devices and CT-based planning. Motion management may comprise either 4D-CT or midbreathing CT image acquisition. Further imaging modalities, such as FDG-PET/CT, may be used when deemed necessary. Gross tumor volumes (GTV) will be contoured and expanded by adequate clinical (CTV) and planning (PTV) safety margins in order to account for subclinical disease and positioning errors. No elective nodal irradiation will be performed. As for organs at risk, both lungs, spinal cord, heart and esophagus must be contoured. In the HYPO-arm, no more than 30% of “both lungs minus GTV” should receive > 20 Gy; in the CON-arm, no more than 35% of “both lungs minus GTV” should receive > 20 Gy.
In the HYPO-arm, 20 fractions of 2.75 Gy will be administered (total dose 55 Gy, corresponding to 70 GyBED, α/β = 10). In the CON-arm, 30 fractions of 2 Gy will be administered (total dose 60 Gy, corresponding to 72 GyBED, α/β = 10). Dose deviations of ±10% are acceptable, when clinically indicated. Radiotherapy is scheduled to start within 72 h after the first administration of durvalumab. Dose prescription will follow international reports (ICRU 50, 62 and 83). Both 3D-conformal and modulated photon radiation techniques, such as IMRT and VMAT/RapdArc, are acceptable. All participating institution are encouraged to perform regular, if no daily, positioning control using either portal imaging or on-board-CT devices.
Study procedures
In order to minimize the risk exposure of patients when subjected to the hypofractionated radiation regimen, a safety lead-in phase with stop-and-go design will precede full enrollment into the HYPO-group (Fig. 2). Toxicity will be evaluated with a 6 + 6 design that is based on the statistical assumption that ≤1 event in n = 18 patients conforms to a non-toxicity scenario, with “event” being defined as the occurrence of pneumonitis grade ≥ 3 within 8 weeks after end of TRT. Consequently, two events in the first six patients or two events in the first 12 or two events in the first 18 patients will result in termination of the HYPO-group (Fig. 2).
During this safety stop-and-go phase, patients will be allocated to treatment arms as follows: While the HYPO-arm is recruiting, patients will exclusively enter this treatment group. During safety evaluation of the six patients of a HYPO-cohort (Stop/ Go decision), patients will be allocated to the CON-group only. If safety criteria in the HYPO-cohort are met, the HYPO-arm will be reopened to continue toxicity assessment, and patients will solely be allocated to this arm (Fig. 1). In order to avoid any bias, patients will be allocated centrally by the study management during this phase. If the non-toxicity criteria in the safety cohorts are met, the study will proceed to the expansion phase, and remaining patients will be randomized into the two treatment arms using an interactive web response system integrated in the electronic case report form (eCRF). A possibly uneven distribution of patients between the treatment groups at this stage will be compensated by a randomization strategy based on the “biased coin” method [15, 16]. In the randomization phase, patients will be stratified according to tumor stage (stage IIIA vs. stage IIIB/IIIC).
In total, 44 patients will be enrolled per group. After n = 18 patients have been enrolled to the HYPO- or CON-treatment arm, respectively, an interim efficacy analysis for the respective arm will be conducted based on the objective response rate (ORR) at 12 weeks after first durvalumab administration. In case of insufficient efficacy of one of the arms (i.e., the number of patients who have achieved a response is eight out of 18 or lower) this treatment arm will be terminated. Recruitment will be halted until results of the interim efficacy analysis are available.
Tumor response will be assessed according to RECIST 1.1 by CT and/ or MRI scans at baseline, 12 weeks after durvalumab initiation and then every 8 weeks. Safety measures will include physical examinations, performance status (ECOG), clinical laboratory profiles and continuous assessments of AEs. An overview of all study procedures is presented in Table 2.
An Independent Safety Monitoring Board (ISMB) will ensure the continued safety of participants throughout the trial. Data management and data quality assurance will be conducted following the Standard Operational Procedures of the Institute of Clinical Cancer Research IKF at Northwest Hospital GmbH (Frankfurt, Germany). An eCRF will be carefully maintained for each participant for data collection, also reporting serious and non-serious AEs following the common criteria for adverse events (CTCAE) version 5.0 throughout the entire trial. After the end of the study, participants will be proactively followed up regarding treatment-related AEs until resolved, returned to baseline or considered irreversible, until lost to follow-up, or withdrawal of study consent. All subjects will be followed for survival. Patients who decline to return to the site for evaluations will be offered a follow-up (FU) by phone every 3 months as an alternative. At the end of the study treatment, the investigators are responsible for the further treatment of the patient and must ensure that he or she receives appropriate standard of care or other appropriate therapies.
Sampling for translational research
If patients participate in the translational research program, blood samples will be collected prior to cycles 1, 2 and 4 and at the time of disease progression (or end of treatment, EOT) along with stool samples (Table 2). Samples of untreated tumor lesions serving as baseline specimens will be collected as paraffin-embedded tissue. If re-biopsies are taken at the time of progression, samples should also be also submitted for translational research.
Study endpoints
The primary endpoint of the study will be toxicity, defined by the occurrence of treatment-related pneumonitis grade ≥ 3. The ORR evaluated 12 weeks after first durvalumab administration (according to RECIST 1.1) is set as the primary efficacy endpoint. Secondary endpoints of the study comprise the occurrence of treatment-related AEs and serious AEs (SAEs), frequency of prespecified abnormal laboratory parameters, progression-free survival (PFS) and duration of clinical benefit, metastasis-free survival, overall survival (OS), and QoL.
Patient vulnerability and its association with survival and outcome will be assessed as an exploratory endpoint. To this end, the G8-screening questionnaire, a simple and fast screening tool for identifying geriatric risk profiles with a strong prognostic value for functional decline and OS in older populations with cancer, will be used [17]. Furthermore, biomaterials will be collected during the trial for correlation analyses on selected molecular parameters and clinical data in order to identify molecular biomarkers predictive for response to therapy. This approach is deemed appropriate to obtain hypothesis-generating data for future research due to the explorative character of this trial.
Statistical analysis
Sample size justification
Safety run-in phase (HYPO-group)
With regard to the pneumonitis grade ≥ 3 rate, this phase is designed to distinguish between a toxicity scenario (pTox = 0.15) and a non-toxicity scenario (pTox = 0.035). A sample size of n = 18 will yield a probability of 0.78 to correctly detect the toxicity scenario, while the non-toxicity scenario will correctly be detected with a probability of 0.87. These probabilities are based on the decision rule that, if the number of patients with pneumonitis grade ≥ 3 in this cohort is ≥2, recruitment to the HYPO-group will be terminated.
Interim efficacy analysis regarding ORR and expansion phase
An interim efficacy analysis based on the ORR will be conducted after n = 18 patients in each arm have completed radiotherapy and the 18th patient has undergone first radiographic assessment (at 12 weeks after first durvalumab dose).
Previously, an ORR of 45% after radiotherapy alone has been reported in a Japanese population of elderly patients with unresectable stage III NSCLC [18]. Based on this and the observation that Asian ethnicity is associated with a favorable prognosis, we assume for the TRADE-hypo trial that an ORR higher than 0.42 in both treatment arms can be demonstrated, i.e. the null hypotheses for arm HYPO and CON are defined as H0 HYPO: π HYPO ≤ 0.42 and H0 CON: π CON ≤ 0.42, where π HYPO and π CON denote the actual ORR in arm HYPO and CON, respectively [19, 20]. Under the alternative hypothesis, it is assumed that both π HYPO and π CON amount to 0.60. Using an optimal Simon’s two-stage design with a one-sided significance level of α = 0.10 and a power of 1-β = 0.80 for each hypothesis test, n = 40 patients per arm are required, while the interim analysis is conducted after n = 18 patients per arm have been recruited to the trial. After successfully passing the safety analysis in the HYPO-group, if among 18 patients in the HYPO- or CON-arm, the number of patients who have achieved a response is eight or lower, the respective arm will be closed. Otherwise, an additional number of 22 patients will be enrolled into the respective arm. The null hypothesis of the respective arm can be rejected if at least 21 of all 40 patients per arm achieve a response. Sample size calculation was done using the R package “OneArmPhaseTwoStudy” [21].
To account for an estimated dropout rate of 10%, four patients will additionally be recruited to each treatment arm. Deviations from planned sample sizes will be handled as described by Englert & Kieser, allowing strict control of the aspired type I error rate in each arm [22].
Methods of statistical analysis
The primary population for evaluating all efficacy endpoints and subject characteristics is defined as all patients enrolled according to initial allocation/randomization (intention-to-treat population, IIT). Secondary efficacy analyses will be carried out on the per-protocol (PP) population. The safety population comprising all patients who received at least one dose of the study medication will be used for all safety endpoints and will be analyzed according to the actual treatment received.
Response rates with confidence intervals (CI) and p-values in both study arms will be estimated taking into account the two-stage nature of the design [23, 24]. Secondary endpoints will be evaluated descriptively. All toxicities will be summarized by relative and absolute frequency, and severity grade based on CTCAE V5.0. AE and SAE summary tables will provide the number and percentage of patients with AEs and the 95% Clopper-Pearson type CIs. All analyses will be done using SAS version 9.4 (SAS Institute, Cary, NC) or higher.
Trial status
As of July 15th 2020, eight study sites are initiated. First initiation coincided with the beginning of the Covid-19 pandemic in Germany. Therefore, recruitment of patients was withheld. On May 8th 2020, recruitment was resumed after consultation with the ISMB. The first patient was enrolled on July 13th 2020.
Discussion
In recent years, the concept of restoring the patients’ antitumor immunity by immune checkpoint inhibition has revolutionized cancer therapy, especially in advanced melanoma, renal carcinoma and NSCLC. Immune checkpoint molecules efficiently regulate T cell activation, and thus enable tumor cells to evade the immune system, for example by hijacking the PD-1/ PD-L1 interaction to downregulate effector T cells [25, 26]. To date, several human monoclonal antibodies pharmacologically blocking these interactions have been implemented in cancer therapy, such as the anti-PD-1 antibody pembrolizumab that has been approved in combination with chemotherapy for non-squamous NSCLC, irrespective of PD-L1 expression [27, 28].
Several studies have shown that immune checkpoint inhibition and radiotherapy in combination can act synergistically to further boost antineoplastic effects [29,30,31,32]. Although in a large phase III trial, no benefit of blockade of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) after radiotherapy was observed in metastatic prostate cancer [33], clinical trials, such as NICOLAS (NCT02434081) and DETERRED (NCT02525757), investigating concurrent PD-(L)1-directed immunotherapy and chemoradiotherapy in patients with locally advanced lung cancer, have confirmed efficiency at modest toxicity rates, particularly with respect to pneumonitis, which is one of the most threatening complications in NSCLC patients [34,35,36]. The PACIFIC trial showed an impressive improvement regarding PFS and OS with manageable side effects with durvalumab treatment after photon radiotherapy combined with chemotherapy in stage III, unresectable NSCLC patients, which has resulted in the approval of durvalumab consolidation after chemoradiotherapy [8, 9]. Safety of durvalumab in combination with concurrent palliative radiotherapy has also been confirmed in a smaller series of heterogeneous metastatic tumor patients [37]. Further, the combination may also prove to be an effective therapeutic concept in other entities, as for example is currently investigated in patients with head-and-neck cancer (NCT03283605) [38].
With regard to elderly and frail patients with unresectable stage III NSCLC unfit for chemotherapy, we hypothesize that durvalumab in combination with mildly hypofractionated TRT is safe and effective, given that feasibility and activity of this regime have been demonstrated in combination with chemotherapy in stage III patients [39, 40]. To date, no prospective trial has investigated such a therapeutic approach. Taking into account the concern of a cumulative risk for severe pneumonitis from applying both TRT and immunotherapy, the TRADE-hypo trial will investigate two radiation regimens combining durvalumab therapy with either conventionally fractionated or hypofractionated TRT.
The study design includes a safety stop-and-go phase preceding full enrollment to ensure that the hypofractionated treatment regimen can instantly be discontinued if deemed unsafe. Taking this and the interim efficacy analysis into account, this study is designed to efficiently avoid inadequate therapy and unnecessary costs. On the other hand, it may reveal a safe and efficient first-line immunoradiotherapeutic strategy for frail and elderly NSCLC stage III patients unable to receive chemotherapy, and, thus, provide an additional and optimized therapeutic option for this potentially undertreated patient cohort. Furthermore, data from this study may guide the design of larger, randomized trials investigating such an approach in this context.
In the accompanying translational research project, the host-tumor biology will be analyzed with a focus on immunological mechanisms responsible for antitumoral effects of both checkpoint inhibitors and radiotherapy, in an attempt to identify biomarkers predictive of therapy response. Eventually, this study may provide valuable data to explore how antitumoral immunological mechanisms may be activated in non-responders, currently representing about half the patients treated with immune-checkpoint inhibitors [27, 28, 41].
Availability of data and materials
Data generated by this study will be available for access from the corresponding author upon reasonable request.
Change history
10 August 2023
A Correction to this paper has been published: https://doi.org/10.1186/s12885-023-11270-x
Abbreviations
- AE:
-
Adverse event
- CCI:
-
Charlson comorbidity index
- CI:
-
Confidence interval
- CT:
-
Computed tomography
- CTCAE:
-
Common Terminology Criteria for Adverse Event
- CTLA-4:
-
Cytotoxic T lymphocyte-associated antigen 4
- CTV:
-
Clinical target volume
- ECG:
-
Electrocardiogram
- ECOG:
-
Eastern Cooperative Oncology Group
- eCRF:
-
Electronic case report form
- EOT:
-
End of treatment
- FU:
-
Follow-up
- GTV:
-
Gross tumor volume
- Gy:
-
Gray
- IP:
-
Investigational Product
- ISMB:
-
Independent Safety Monitoring Board
- ITT:
-
Intent-to-treat
- MRI:
-
Magnetic resonance imaging
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PD-1:
-
Programmed cell death 1
- PD-L1:
-
Programmed cell death ligand 1
- PFS:
-
Progression-free survival
- PP:
-
Per-protocol
- PTV:
-
Planning target volume
- QoL:
-
Quality of life
- RECIST 1.1:
-
Response Evaluation Criteria in Solid Tumors, version 1.1
- SAE:
-
Serious adverse event
- TRT:
-
Thoracic radiation therapy
References
Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, Mok TS, Reck M, Van Schil PE, Hellmann MD, et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv192–237.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30.
Howlader NNAKM, et al. SEER Cancer statistics review, 1975–2010. Bethesda: National Cancer Institute; 2013.
Miller ED, Fisher JL, Haglund KE, Grecula JC, Xu-Welliver M, Bertino EM, He K, Shields PG, Carbone DP, Williams TM, et al. Identifying patterns of care for elderly patients with non-surgically treated stage III non-small cell lung cancer: an analysis of the national cancer database. Radiat Oncol. 2018;13(1):196.
Driessen EJM, Schulkes KJG, Dingemans AC, van Loon JGM, Hamaker ME, Aarts MJ, Janssen-Heijnen MLG. Patterns of treatment and survival among older patients with stage III non-small cell lung cancer. Lung Cancer. 2018;116:55–61.
Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10(7):718–26.
Ko EC, Raben D, Formenti SC. The integration of radiotherapy with immunotherapy for the treatment of non-small cell lung Cancer. Clin Cancer Res. 2018;24(23):5792–806.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Yokoi T, Chiappori A, Lee KH, de Wit M, et al. Durvalumab after Chemoradiotherapy in stage III non-small-cell lung Cancer. N Engl J Med. 2017;377(20):1919–29.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Kurata T, Chiappori A, Lee KH, de Wit M, et al. Overall survival with Durvalumab after Chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379(24):2342–50.
Stinchcombe TE, Zhang Y, Vokes EE, Schiller JH, Bradley JD, Kelly K, Curran WJ Jr, Schild SE, Movsas B, Clamon G, et al. Pooled analysis of individual patient data on concurrent Chemoradiotherapy for stage III non-small-cell lung Cancer in elderly patients compared with younger patients who participated in US National Cancer Institute cooperative group studies. J Clin Oncol. 2017;35(25):2885–92.
Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing tumor immunity with fractionated radiation. Int J Radiat Oncol Biol Phys. 2012;83(4):1306–10.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15(17):5379–88.
Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174(12):7516–23.
Marconi R, Strolin S, Bossi G, Strigari L. A meta-analysis of the abscopal effect in preclinical models: is the biologically effective dose a relevant physical trigger? PLoS One. 2017;12(2):e0171559.
Smith R. Sequential treatment allocation using biased coin designs. J R Stat Soc Ser B Methodol. 1984;46(3):519–43.
Frane J. A method of biased coin randomization, its implementation, and its validation. Drug Information J. 1998;32(2):423–32.
Kenis C, Decoster L, Van Puyvelde K, De Greve J, Conings G, Milisen K, Flamaing J, Lobelle JP, Wildiers H. Performance of two geriatric screening tools in older patients with cancer. J Clin Oncol. 2014;32(1):19–26.
Atagi S, Kawahara M, Yokoyama A, Okamoto H, Yamamoto N, Ohe Y, Sawa T, Ishikura S, Shibata T, Fukuda H, et al. Thoracic radiotherapy with or without daily low-dose carboplatin in elderly patients with non-small-cell lung cancer: a randomised, controlled, phase 3 trial by the Japan clinical oncology group (JCOG0301). Lancet Oncol. 2012;13(7):671–8.
Ou SH, Ziogas A, Zell JA. Asian ethnicity is a favorable prognostic factor for overall survival in non-small cell lung cancer (NSCLC) and is independent of smoking status. J Thorac Oncol. 2009;4(9):1083–93.
Zhou W, Christiani DC. East meets west: ethnic differences in epidemiology and clinical behaviors of lung cancer between east Asians and Caucasians. Chin J Cancer. 2011;30(5):287–92.
Kieser MWM, Englert S, Kunz CU, Rauch G. OneArmPhaseTwoStudy: an R package for planning, conducting, and analysing single-arm phase II studies. J Stat Soft. 2017;81:1–28.
Englert S, Kieser M. Methods for proper handling of overrunning and underrunning in phase II designs for oncology trials. Stat Med. 2015;34(13):2128–37.
Kunzmann K, Kieser M. Point estimation and p-values in phase II adaptive two-stage designs with a binary endpoint. Stat Med. 2017;36(6):971–84.
Kunzmann K, Kieser M. Test-compatible confidence intervals for adaptive two-stage single-arm designs with binary endpoint. Biom J. 2018;60(1):196–206.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800.
Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99(19):12293–7.
Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung Cancer. N Engl J Med. 2016;375(19):1823–33.
Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung Cancer. N Engl J Med. 2018;378(22):2078–92.
Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, Garon EB, Lee P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017;18(7):895–903.
Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJ, Stratford IJ, Poon E, Morrow M, Stewart R, et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014;74(19):5458–68.
Golden EB, Chhabra A, Chachoua A, Adams S, Donach M, Fenton-Kerimian M, Friedman K, Ponzo F, Babb JS, Goldberg J, et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol. 2015;16(7):795–803.
Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. 2018;18(5):313–22.
Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–12.
Peters S, Felip E, Dafni U, Belka C, Guckenberger M, Irigoyen A, Nadal E, Becker A, Vees H, Pless M, et al. Safety evaluation of nivolumab added concurrently to radiotherapy in a standard first line chemo-radiotherapy regimen in stage III non-small cell lung cancer-the ETOP NICOLAS trial. Lung Cancer. 2019;133:83–7.
Lin SH, Lin Y, Yao L, Kalhor N, Carter BW, Altan M, Blumenschein G, Byers LA, Fossella F, Gibbons DL, et al. Phase II trial of concurrent Atezolizumab with Chemoradiation for Unresectable NSCLC. J Thorac Oncol. 2020;15(2):248–57.
Belluomini L, Fiorica F, Frassoldati A. Immune checkpoint inhibitors and radiotherapy in NSCLC patients: not just a fluke. Oncol Ther. 2019;7(1):83–91.
Levy A, Massard C, Soria JC, Deutsch E. Concurrent irradiation with the anti-programmed cell death ligand-1 immune checkpoint blocker durvalumab: single Centre subset analysis from a phase 1/2 trial. Eur J Cancer. 2016;68:156–62.
Bahig H, Aubin F, Stagg J, Gologan O, Ballivy O, Bissada E, Nguyen-Tan FP, Soulieres D, Guertin L, Filion E, et al. Phase I/II trial of Durvalumab plus Tremelimumab and stereotactic body radiotherapy for metastatic head and neck carcinoma. BMC Cancer. 2019;19(1):68.
Maguire J, Khan I, McMenemin R, O'Rourke N, McNee S, Kelly V, Peedell C, Snee M. SOCCAR: a randomised phase II trial comparing sequential versus concurrent chemotherapy and radical hypofractionated radiotherapy in patients with inoperable stage III non-small cell lung Cancer and good performance status. Eur J Cancer. 2014;50(17):2939–49.
Iqbal MS, Vashisht G, Mulvenna P, McDonald F, Turnbull H, Atherton P, Bradshaw A, Simmons T, Kovarik J, Singhal S, et al. Hypofractionated concurrent chemoradiation in non-small cell lung cancer (NSCLC): efficacy and toxicity of the SOCCAR trial regime in real world practice. Lung Cancer. 2018;115:S64.
Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, Ready NE, Gerber DE, Chow LQ, Juergens RA, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18(1):31–41.
Acknowledgements
The authors would like to thank the members of the Independent Safety Monitoring Board. This trial is conducted in cooperation with the Young Medical Oncologists Group and the Thoracic Oncology Working Group of the Arbeitsgemeinschaft Internistische Onkologie (AIO) within the German Cancer Society. We acknowledge financial support by the Baden-Württemberg Ministry of Science, Research and the Arts and by the Ruprecht-Karls-University Heidelberg.
Funding
The Institute of Clinical Cancer Research IKF at Northwest Hospital GmbH (Steinbacher Hohl 2–26, D-60488 Frankfurt a.M.) acts as the legal sponsor and finances the trial. Financial support and drug supply is granted by the pharmaceutical company AstraZeneca/ MedImmune. Open access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
FB, IC, MT and SR developed the study idea, wrote the protocol and coordinated funding, regulatory permission and approval processes. FB is lead investigator of this study; SR is deputy lead investigator and will supervise all radiation treatments. JK calculated (bio-) statistical models for sample size, study hypothesis, and endpoint determinations. PC and FB developed biomarker evaluation strategies and MS will coordinate sample collection and processing during this study. LB participated in writing the manuscript. DM is representative of the Institute of Clinical Cancer Research IKF at Northwest Hospital GmbH that is legal sponsor of the study, gave advice on study design and is responsible for study management and logistics. All authors have read and approved the current version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval by the Ethics Committee of the Medical Faculty of the University of Heidelberg was obtained (AFmu-683/2019 March 18, 2020). Before participation in the clinical study, written informed consent will be obtained from each subject. Furthermore, the DEGRO expert panel reviewed the radiation regimen to rule out that permission of radiation administered would have to be requested from the BfS (Federal office of radiation protection; “Anfrage 167 / 09 July, 2019”). Additionally, the Paul Ehrlich Institute (competent authority for approval of clinical trials using medicinal products for human use in Germany) approved the study (no. 3881/02, March 10, 2020).
Consent for publication
Not applicable, as no individual patient data are contained in this manuscript.
Competing interests
The TRADE-hypo trial receives funding from AstraZeneca/ MedImmune. However, AstraZeneca/ MedImmune has not been involved in study design, data collection, management, data analysis and interpretation, or in the decision to submit this protocol for publication. All authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bozorgmehr, F., Chung, I., Christopoulos, P. et al. Thoracic radiotherapy plus Durvalumab in elderly and/or frail NSCLC stage III patients unfit for chemotherapy - employing optimized (hypofractionated) radiotherapy to foster durvalumab efficacy: study protocol of the TRADE-hypo trial. BMC Cancer 20, 806 (2020). https://doi.org/10.1186/s12885-020-07264-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-07264-8