
Abstract
Background
HOXA family genes were crucial transcription factors involving cell proliferation and apoptosis. While few studies have focused on HOXA10 in AML. We aimed to investigate the prognostic significance of HOXA10.
Methods
We downloaded datasets from GEO and BeatAML database, to compare HOXA expression level between AML patients and controls. Kaplan-Meier curves were used to estimate the impact of HOXA10 expression on AML survival. The differentially expressed genes, miRNAs, lncRNAs and methylated regions between HOXA10-high and -low groups were obtained using R (version 3.6.0). Accordingly, the gene set enrichment analysis (GSEA) was accomplished using MSigDB database. Moreover, the regulatory TFs/microRNAs/lncRNAs of HOXA10 were identified. A LASSO-Cox model fitted OS to clinical and HOXA10-associated genetic variables by glmnet package.
Results
HOXA10 was overexpressed in AML patients than that in controls. The HOXA10-high group is significantly associated with shorter OS and DFS. A total of 1219 DEGs, 131 DEmiRs, 282 DElncRs were identified to be associated with HOXA10. GSEA revealed that 12 suppressed and 3 activated pathways in HOXA10-high group. Furthermore, the integrated regulatory network targeting HOXA10 was established. The LASSO-Cox model fitted OS to AML-survival risk scores, which included age, race, molecular risk, expression of IKZF2/LINC00649/LINC00839/FENDRR and has-miR-424-5p. The time dependent ROC indicated a satisfying AUC (1-year AUC 0.839, 3-year AUC 0.871 and 5-year AUC 0.813).
Conclusions
Our study identified HOXA10 overexpression as an adverse prognostic factor for AML. The LASSO-COX regression analysis revealed novel prediction model of OS with superior diagnostic utility.
Similar content being viewed by others


Background
Acute myeloid leukemia (AML) accounts for 80% of acute leukemia in adults. AML is characterized by unlimited clonal proliferation and accumulation of myeloid progenitors [1]. The 5-year survival for AML patients is no more than 50%, which is less than 20% in elderly AML patients [2]. To estimate risk and survival of AML patients, quite a few prediction models have been developed. European LeukemiaNet (ELN) 2017 risk stratification is the most commonly used risk model, which stratified AML patients based on recurrent cytogenetics and molecular mutations abnormalities [3]. A comprehensive evaluation of genetic variables is crucial for risk stratification and will guide treatment decisions. Other traditional prognostic factors include age, WBC count, LDH level etc. [4]. Whereas the biomarkers with prognostic value are still being explored to improve the risk model for AML.
The genetic alterations of transcription factors (TFs) occur frequently in AML, which exert important effects in pathogenetic process and associate with prognosis [5,6,7,8]. HOXA (homeobox protein HOX cluster A) family genes, encoding highly conserve TFs with DNA binding homeobox binding motifs, play a crucial role in adult hematopoiesis [9,10,11], while aberrant overexpression of HOXA promotes oncogenesis [12]. The previous studies indicated that genetic alterations in AML resulted in HOXA overexpression, such as KMT2A rearrangement [13], FLT-ITD [13], MLL gene abnormality [14,15,16,17]. HOXA9, HOXA7, HOXA11 were associated with adverse prognosis in AML [18, 19]. Overexpression of HOXA10 was reported to interrupt hematopoietic process [20] and lead to myeloid leukemogenesis in mice model [21, 22]. While the prognostic significance of HOXA10 for AML has been rarely explored.
Nowadays, multidimensional information has been accumulated for AML other than gene mutations and karyotypes, including gene expression, non-coding RNA profile, gene methylation profile, copy number variation, etc. In the present study, we explored regulatory genetic or epigenetic variables of HOXA10, such as methylation of CpG, copy number variation (CNV), lncRNA, microRNA and TF, which affect the gene expression. The interaction of lncRNA and miRNA sponges form competing endogenous RNA (ceRNA) network regulating gene expression and pathways. Then Lasso-Cox model was used to fit AML survival to prediction model, including clinical features and HOXA10-associated genetic/epigenetic variables. Our work offered evidence for using HOXA10 as a prognostic marker for AML, and establishment of novel risk model to predict AML survival.
Methods
Data source
We downloaded the microarray data of GSE15061 [23] (202 AML and 69 controls, Affymetrix U133 Plus 2.0 Array), GSE30029 [24] (46 AML and 31 controls, Illumina HumanHT-12 V3.0 beadchip), GSE114868 [25] (194 AML and 20 controls, Affymetrix Transcriptome Array 2.0), GSE13159 [26] (501 AML and 73 controls, Affymetrix U133 Plus 2.0 Array) form GEO database (https://www.ncbi.nlm.nih.gov/geo/). The RNA-seq data was obtained from BeatAML database [27] (474 AML and 33 controls, http,//www.vizome.org/aml). For micro-array data, expression level of a gene was calculated as the mean value (M value) of all probe sets annotating to it. For RNA-seq data from BeatAML database, the log2 transformed Reads Per Kilobase Million (RPKM) data was utilized.
Comparison of HOXA10 expression level between AML patients and controls
The 5 public micro-array/RNA-seq datasets were used to compare HOXA10 expression level between AML patients and controls. GSE30029 dataset sampled from CD34+ bone marrow cells, while other datasets were obtained from unsorted bone marrow cells. The micro-array and RNAseq data were normalized before analysis by R program (3.6.0). Then expression level of HOXA10 was compared between AML and controls, using unpaired t-test.
Kaplan-Meier analysis of HOXA10 on AML survival
The RNAseq data (count) of 151 AML patients was download for AML cohort from TCGA database (https://portal.gdc.cancer.gov/). The median RPKM was employed to divide the patients into HOXA10-high and HOXA10-low groups. Kaplan-Meier plot and logrank test were used to compare OS and DFS between 2 groups.
Genome-wide gene/miRNA/lncRNA expression profiles associated with HOXA10
The differentially expressed genes/miRNAs/lncRNAs (DEGs/DEmiRs/DElncRs), between HOXA10-high and -low groups, were identified by “DESeq2” package and R (version 3.6.0). Then we accessed the HOXA10-associated cell signaling pathways using gene set enrichment analysis (GSEA) based on MSigDB database (http://software.broadinstitute.org/gsea/msigdb) [28,29,30].
Overrepresentation analysis of aberrantly expressed genes associated with HOXA10
To demonstrate the implication of DEGs, the ClueGO plugin of Cytoscape software (version 3.7.3) was employed to perform functional enrichment analysis based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome pathway databases. Gene ontology (GO) based functional enrichment was conducted by the “topGO” package (R 3.6.0, Bioconductor 3.10), and summarized by the “REVIGO” package. The enriched GO/KEGG/REACTOME terms or pathways were defined to be significant with an adjusted p value < 0.05.
Establishment of upstream regulatory network
The upstream TFs/miRNAs/lncRNAs, targeting HOXA10 gene, were identified, based on TF/microRNA/lncRNA target predicting algorithm and correlating analysis, using Gene Transcription Regulation Database (GTRD, http://gtrd.biouml.org/) [31], miRWalk 2.0 (http://zmf.umm.uni-heidelberg.de/) [30, 32] and prediction module of lncBase v2 [33] online tools. Then combined with the results of genome-wide expression analysis, we identified the overlapped genes/miRNAs/lncRNAs, which were predicted to target HOXA10 and differentially expressed.
Establishment of prediction model for AML survival
In order to improve the prognostic model, a comprehensive survival analysis was performed, which integrated clinical features, HOXA10 expression, methylation, CNV and upstream TFs/microRNAs/lncRNAs expression. RNA-seq data (counts, IlluminaHiSeq), miRNAseq data (RPM value, illuminaGA), beta value of methylation (HumanMethylation450), and gene-level copy number data (GISTIC 2 method) regarding HOXA10 were downloaded from the TCGA database (https://portal.gdc.cancer.gov/). Since AML-M3 patients have a distinct prognostic profile in comparison with other subtypes, we excluded such patients. The traditional prognostic factors were brought into analysis, including age, gender, race, risk stratification of cytogenetics, risk stratification of molecular mutations and WBC count. The molecular mutations and cytogenetic risk stratification were based on ELN2017 recommendations [3], which classified AML patients into good/intermediate/poor groups based on molecular mutations and cytogenetics. Finally, we screened and included113 AML patients with all abovementioned information. The OS and included factors were fitted to least absolute shrinkage and selection operator (LASSO) -Cox model, resulting in prediction model for AML survival.
Statistical analysis
We established a LASSO-Cox regression model [34], using glmnet package and R program (3.6.0). LASSO method for variable selection penalizes the data fitting criteria, which gets rid of less informative predicting variables to reduce complexity and maker models more interpretable. For each of the LASSO-screened variables, the final coefficients were the average estimates of the coefficients obtained from cross-validation development. To evaluate the diagnostic utility of prediction model, time dependent receiver operating characteristic (ROC) curve was used and 1-year/3-year/5 year area under curve (AUC) were calculated. Wilcoxon rank-sum was employed for comparisons of continuous variables between subgroups. Chi-square tests were used to test the association of categorical variables.
Results
Overexpression of HOXA10 in AML
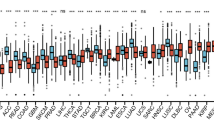
The higher expression level of HOXA10 was revealed in AML patients than that in control group, in unsorted and CD34+ bone marrow cells (Fig. 1a-e). HOXA10 expression signature was similar across different cell subpopulations of AML. Furthermore, results from GSE13159 implicated that the HOXA10 expression was higher in AML than that in other myeloid neoplasms (MDS/CML) and lymphoid malignancies (T-ALL/B-ALL), which indicated that HOXA10 overexpression may be AML-specific signature.

Comparison of expression level in AML patients and controls using data derived from GSE15061(a), GSE30029(b), GSE114868(c), GSE13159(d) and BeatAML(e). The expression level of HOXA10 was represented by log2 transformed normalized mean (M) value or RPKM value. Underlying difference of HOXA10 expression was revealed between AML and healthy control group by all datasets (a-e), and the difference between AML and other myeloid malignancy (MDS, CML) was uncovered by GSE13159 (Fig. d). The conclusion was robust no matter the expression data was derived from unsorted bone marrow or CD34+ cells
High expression of HOXA10 is associated with adverse prognosis
The clinical characteristics of HOXA10-high and -low groups were shown in Table 1. The proportion of AML-M3 is higher in HOXA10-low group than that in HOXA10-high group (p < 0.0001). HOXA10-high AML patients were associated more advanced ELN2017 risk stratification (p < 0.0001).
The Kaplan-Meier plots indicated that the AML survival of HOXA10-high group was significantly shorter than that of HOXA10-low group (Fig. 2a&b). Median OS of HOXA10-high and -low groups are 12.105 vs 24.210 months respectively, and logrank p value is 0.0302. The median DFS of HOXA10-high and low are 11.809 vs 28.389 months respectively, and logrank p value is 0.0207. The results indicated that HOXA10 overexpression is an adverse prognostic factor for AML patients.

The Kaplan-Meier plots of HOXA10 expression for DFS (a) and OS (b). The survival (DFS and OS) of HOXA10-low AML group is twice as longer as that of HOXA10-high group
Genome-wide gene/miRNA/lncRNA profiles associated with HOXA10 expression
1219 DEGs, 131 DEmiRs and 282 DElncRs were identified by comparing HOXA10-high and HOXA10-low AML groups from TCGA (Fig. 3a). The heatmap for top DEGs was shown in Fig. 3b, in which the top differentially expressed genes, filtered by adjusted p value, were revealed between HOXA10-high and low groups. Wnt Family Member 7B (WNT7B), Neuregulin 4 (NRG4) and HOXA11 were the top DEG, which were overexpressed in HOXA10-high groups in comparison with that in HOXA10-low group. Other HOX family genes, like HOXA2/HOXA3/HOXA4/HOXA5/HOXA6/HOXA7/HOXB6/HOXB8/HOXB9, were upregulated correlating to HOXA10 expression. The distinct expression signature between 2 groups may help us to investigate and uncover potential biomarkers.

The volcano plot (a) and heatmap (b) showed the distribution and top DEGs. 1219 DEGs were reveal by comparing the gene expression level between HOXA10-high and low group, with 433 upregulated DEGs and 786 downregulated DEGs. The heatmap showed the distinct expression signature of HOXA10-high (left columns) and low groups (right columns), displaying top differentially expressed genes screened by adjusted P value. The over-expressed genes were colored in red, while the under-expressed genes were in blue. The dotplot of GSEA results and barplot of ORA results were shown in c and d, respectively. In c, the significantly activated and suppressed pathways derived from GSEA were listed, in which the color of dots represent the adjusted P value and the diameter stand for enriched gene count. In d, the top enriched pathways obtained from GO/KEGG/Reactome analysis were shown, while the X axis stands for -ln(q value)
The results of GSEA indicated 12 suppressed cell pathways and 3 activated pathways were significantly correlated with HOXA10 expression (Fig. 3c & Supplementary Table 1). The suppressed pathways in HOXA10-overexpressed patients included PI3K-Akt signaling, hematopoietic cell lineage, cAMP signaling, etc. while the activated pathways included ribosome, oxidative phosphorylation, lysosome, etc.
Overrepresentation analysis for DEGs
GO analysis revealed that DEGs were significantly enriched in the following biological processes (BP): cell-cell signaling, cell communication, etc. Cell component (CC) analysis revealed that DEGs were predominantly located in the plasma membrane region, extracellular matrix, etc. Molecular mutations function (MF) analysis demonstrated that DEGs were enriched in signaling receptor activity, extracellular matrix structural constituent, etc. For KEGG pathway analysis, DEGs were significantly enriched in the following pathways: PI3K-Akt signaling pathway, Ras signaling pathway, etc. In Reactome analysis, the DEGs were enriched in signaling by PDGF, signaling by ROBO receptor, etc. The detail ORA (overrepresentation analysis) results are listed in Supplementary Table 2, and top enriched pathways are shown in Fig. 3d.
Genome-wide methylation profile associated with HOXA10 expression
A total of 76 DMRs within exonic regions were uncovered. The detail DMRs within exonic regions were listed in Supplementary Table 3. The methylation level of HOXA10 was significantly negatively correlated with HOXA expression. Among the methylated CpG sites of HOXA10, cg21172377 is significantly differentially methylated along with HOXA10 expression. And the AML patients with hypermethylated cg21172377 have significantly shorter OS, according to survival analysis using MethSurv [35] (https://biit.cs.ut.ee/methsurv/). Therefore, beta value of cg21172377 was used to represent the methylation level of HOXA10 in the following analysis.
Regulatory network of HOXA10
The upstream ceRNA network and TFs regulating HOXA10 were shown in Fig. 4. The HOXA10 related TFs/miRNAs/lncRNAs included BCL6B, NR2F2, KLF1, ZSCAN4, IKZF2, LINC00649, LINC00839, LINC00707, HOXA11-AS, FENDRR, miR-424-5p, miR-130a-3p, miR-497-5p and miR-195-5p, all of which are predicted by miRWalk 2.0 and lncBase v2 online tools and differentially expressed between HOXA10-high and -low group.

The regulatory network of HOXA10 expression consisting related TFs, lncRNAs, microRNAs, which were then inputted as variables in LASSO analysis to reveal the prediction model
Identification of key prognostic markers
LASSO regression analysis identified the OS fitted predictive model, including age, race, molecular risk, expression level of IKZF2/LINC00649/LINC00839/FENDRR/has-miR-424-5p. The developing model process identified AML-survival risk scores (ARS) to calculate each patient using variables weighted by coefficients (Table 2). The cut-off value was calculated by “cutoffROC” package (Fig. 5d), which is 1.315. Time dependent ROC was performed using survivalROC package, and the 1-year/3-year/5-year AUC are 0.808/0.839/0.786 respectively, implicating the diagnostic utility is satisfying.

a showed the distribution of risk score calculated by ARS model in 113 AML patients, with 1.32 as the cutoff value. b displayed the survival events distribution, in which the dotted line separated the high and low risk groups. Risk to variable heatmap (c) revealed the top variables that distributed differently between high and low risk group. Time dependent ROC curves of ARS model showing 1-year/3-year/5-year AUC in c, showing the diagnostic utility
By the cut-off ARS value, the AML cohort was classified as high-risk and low-risk groups. The ARS distribution plot, survival-events plot and risk-to-variable heatmap were shown in Fig. 5a&b&c. Then we used Kaplan-Meier plot to analysis the survival difference between ARS-high risk and low risk group (Fig. 6, logrank p < 0.0001, HR = 27.66). The median OS of ARS-low patients is 28.373 months and that of ARS high patients is only 3.987 months. The performance of ARS model is encouraging, but further prospective studies are needed to evaluate the predictive value of this model.

The Kaplan-Meier plot comparing the overall survival of ARS-high and ARS-low groups. Notably, the survival of ARS-high group is far more inferior
Discussion
HOXA family genes are well known as the crucial transcription factors in pathogenesis and development of AML. HOXA10 belongs to the HOXA gene superfamily, dysregulation of which has been observed in several solid tumors [36,37,38]. HOXA10 plays a role in myeloid differentiation, leukemogenesis and chemoresistance in AML [39,40,41]. In the present study, we investigated the prognostic significance of HOXA10 in AML, which has been rarely described and reported previously. The results of expression analysis demonstrated that AML patients have aberrant HOXA10 expression in comparison with controls. High HOXA10 expression level is significantly associated with worse OS and DFS of AML, based on Kaplan-Meier curve and logrank test. Therefore, HOXA10 may serve as a prognostic marker for AML patients.
To explore the underlying enriched pathways of DEG, the ORA was performed. In the GO part of our ORA, we uncovered that the biological processes of DEG were enriched in cell-cell signaling and cell communication, and the cell components were enriched in plasma cell membrane and extracellular matrix. Correspondingly, the molecular functions were enriched in signaling receptor activity and extracellular matrix structural constituent. To explore the detail cell-cell signaling pathway that the DEGs were involved, the following KEGG and Reactome analysis revealed that the DEGs were enriched in PI3K-Akt signaling, Ras signaling pathway, signaling by PDGF and signaling by ROBO receptor, etc. In addition to activating mutation of NRAS/KRAS in 15–25% AML, the mutations of RAS-regulating genes (NF1 and PTPN11) and RAS-signaling receptor (FLT3 and KIT) are also harbored in AML frequently [42,43,44,45,46]. Hyperactive mutations of NF1 and PTPN11 gene are associated with inferior survival in pediatric and elderly AML [47, 48]. FLT3-ITD mutation is also well-known as a biomarker for worse prognosis in non-APL AML patients [49, 50]. Although NRAS and KRAS genes was not significantly differentially expressed, RAS signaling genes, including PTPN11, FLT3 and KIT, were upregulated significantly in HOXA10-high AML patients. The dysregulation of RAS signaling pathway may lead to unfavorable impact on clinical outcome of HOXA10-high group. PDGF signaling plays a proto-oncogenic role in diverse cancer cells. Imatinib turned out to block PDGF receptor at low dose, exerting a pharmacological effect for BCR-ABL positive CML and FIP1L1-PDGFRA mutated eosinophilic leukemia patients [51, 52], suggesting PDGF signaling as an activated effector in hematological malignancies. Gołos A et al. reported increased ROBO1/2 in AML patients in comparison with normal control, and high ROBO3 expression is associated with cytogenetic high risk and poor prognosis [53]. However, the aberrantly expression pattern of signaling pathway by PDGF/ROBO has not been fully elucidated in AML, which were rarely studied in the prognosis of AML. The enriched pathways obtained by ORA help us to identify expression signature in HOXA10-high group, screen useful biomarkers and provided novel insights into molecular investigation on AML.
Differential expression analysis and GSEA revealed that PI3K-Akt signaling pathway was suppressed associated with HOXA10 overexpression. PI3K-Akt signaling pathway is frequently activated in AML, constitutive activation of PI3K and Akt were found in 50% de novo AML patients [54, 55]. The PI3K-Akt signaling controls leukemic blast cells proliferation and clonogenicity [56, 57]. AML patients with constitutive PI3K-Akt activation have better OS and relapse-free survival [58]. The unfavorable survival profile in HOXA10-high group may attributed to aberrant downregulation of PI3K-Akt signaling.
The results of GSEA demonstrated that ribosome, oxidative phosphorylation, and lysosome pathways were activated in HOXA10-high group. Ribosome pathway is a vital cellular process, and the rate-limiting step of which is the initiation of translation in ribosomes. One of major control factors in the ribosome activity, is EIF2 (eukaryotic initiation factor 2), which is regulated by phosphorylation of α subunit (EIF2α) under diverse stress. Four kinds of EIF2α kinases (EIF2AK1/2/3/4) can affect the activity of EIF2α by phosphorylation of Ser51 [59, 60]. Notably, the expression EIFAK2 and EIFK3 were significantly increased in HOXA10-high group by our results. EIF2AK2 (also named as double-strand RNA-dependent kinase, PKR) responses to various types of stress, including DNA damage, mitochondrial stress, viral infection, growth factor deprivation, cytokines, Toll-like receptor activation and cytotoxic drugs [61,62,63,64,65]. Also EIF2AK2 is the only EIF2α kinase exists in both the cytoplasm and nucleolus, while other 3 kinases present only in the cytoplasm [66]. Cheng X et al. reported that high expression of EIF2AK2 was associated with worse prognosis in AML, and it reduced DNA damage response by inhibiting ataxia-telangiectasia mutated (ATM) activation, leading to accretion of leukemia in mice model [67]. EIF2AK3 (also named as PKR-like endoplasmic reticulum kinase, PERK) is reported to promoted leukemia progress by stimulating the dissemination of leukemia cells in vivo [68]. So, the increased EIF2AK2/3 expression and activated ribosome pathway contributed to the worse outcomes in HOXA10-high patients. The maintenance of leukemia stem cells depends on BCL2 mediated oxidative respiration, instead of glycolysis as in normal hematopoietic cells [69]. The metformin, targeting oxidative phosphorylation (OXPHOS), induces apoptosis of human leukemia cells in an AMPK-independent way [70]. Cytarabine resistant leukemia cells are characterized by activated OXPHOS, with the high level of reactive oxygen species. Additionally, the resistance can be reversed by agents inducing low OXPHOS status [71]. The activation of OXPHOS in HOXA10-high patients may promoted leukemia cell maintenance and chemo-resistance, leading to inferior survival. The biological function of lysosome pathway in AML has not been fully elucidated. While considering lysosome pathway involves in autophagy, which plays a role in leukemic transformation of normal hematopoietic stem cells and chemotherapy response [72], it may be still valuable to explore in this area. The Kaplan-Meier curves confirmed that HOXA10 expression is associated with AML survival, while it didn’t predict OS or DFS significantly in multivariable Cox hazards analysis including other clinical and genetic variables (data not shown), suggesting HOXA10 is not an independent prognostic factor. Since copy number variation (CNV), mutations (not reported in TCGA database), CpG methylation status and upstream TFs/microRNA/lncRNA are the most common gene expression regulators, we investigated whether these factors were associated with HOXA10 expression and established Lasso-Cox model fitting OS to reveal novel prognostic markers. And finally, Lasso-Cox analysis included clinical features, CNV of HOXA10, methylation and expression status of HOXA10, HOXA10 associated TFs/DEmiRs/DElncRs. Due to the obvious correlation and dependency between each variable, the traditional multivariable Cox analysis is of limited utility, where the Lasso-Cox methods showed its superiority. A few prediction models for AML overall survival have been reported (Table 3), including Huang R et al. [73], Mihyang Ha et al. [74], Clinseq-G model [75], ELN2017 recommendation [75], Zejuan Li et al. [75]. The AUC equals to the probability, which a diagnostic classifier will rank a randomly chosen positive instance higher than a negative one, and the highest AUC is established as best practice [76]. Our prediction model has superior AUC than above models, possibly attributing to integrated multidimensional information integrated.
In the ARS model, novel markers are included. IKZF2 is recently found to drive leukemia stem cell renewal and inhibit myeloid differentiation, by regulating HOXA9 and CEBP [77]. The aberrant variation of IKZF2 was also reported in ovarian cancer cell lines [78], adult T cell leukemia [79] and gastric cancer [80], suggesting IKZF2 as a potential oncogenes. The relation of AML and LINC00649 has not been explored. While notably, the relation of HOXA family genes and LINC00649 is very close. Based on Gepia 2.0 (http://gepia2.cancer-pku.cn/), using AML RNAseq dataset of TCGA and Pearson method, LINC00649 is negatively correlated with HOXA1 (p = 0.015), HOXA2 (p = 0.0056), HOXA3 (p = 0.0045), HOXA4 (p = 0.011), HOXA5 (p = 0.0019), HOXA6 (p = 0.0078), HOXA7 (p = 0.00089), HOXA9 (p = 0.002) and HOXA10 (p = 0.0022) significantly. Intriguingly, expression level of all above HOXA genes are significantly associated with AML survival [8], which indicated that LINC00649 targeted genes may exert pathogenetic effect in AML. The investigation of LINC00839, FENDRR and has-miR-424-5p in AML has not been conducted.
The limitation of this model is also obvious, which is lacking prospective large-scale studies for validation. And further experiment is needed to validate the interaction of TFs/microRNAs/lncRNAs with HOXA10.
Conclusions
We identified the overexpression of HOXA10 as an adverse prognostic factor of AML OS and DFS. The novel multidimensional prediction model was established with satisfying diagnostic utility. These results need further clinical and experimental validation.
Availability of data and materials
The data that support the findings of this study are available from GEO database (https://www.ncbi.nlm.nih.gov/geo/), BeatAML database (http://www.vizome.org/aml) and TCGA database (https://portal.gdc.cancer.gov/), which are all publicly available.
Abbreviations
- AML:
-
Acute myeloid leukemia
- ELN:
-
European LeukemiaNet
- CNV:
-
copy number variation
- RPKM:
-
Reads Per Kilobase Million
- HOXA:
-
homeobox protein HOX cluster A
- ceRNA:
-
endogenous RNA
- DEGs/DEmiRs/DElncRs:
-
differentially expressed genes/miRNAs/lncRNAs
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GO:
-
Gene ontology
- LASSO:
-
least absolute shrinkage and selection operator
References
Barbui T, Thiele J, Gisslinger H, Kvasnicka HM, Vannucchi AM, Guglielmelli P, Orazi A, Tefferi A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8(2):15.
Short NJ, Rytting ME, Cortes JE. Acute myeloid leukaemia. Lancet. 2018;392(10147):593–606.
Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47.
Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med. 2015;373(12):1136–52.
Jalili M, Yaghmaie M, Ahmadvand M, Alimoghaddam K, Mousavi SA, Vaezi M, Ghavamzadeh A. Prognostic value of RUNX1 mutations in AML: a meta-analysis. Asian Pac J Cancer Prev. 2018;19(2):325–9.
Tien FM, Hou HA, Tsai CH, Tang JL, Chiu YC, Chen CY, Kuo YY, Tseng MH, Peng YL, Liu MC, et al. GATA2 zinc finger 1 mutations are associated with distinct clinico-biological features and outcomes different from GATA2 zinc finger 2 mutations in adult acute myeloid leukemia. Blood Cancer J. 2018;8(9):87.
Taketani T, Taki T, Sako M, Ishii T, Yamaguchi S, Hayashi Y. MNX1-ETV6 fusion gene in an acute megakaryoblastic leukemia and expression of the MNX1 gene in leukemia and normal B cell lines. Cancer Genet Cytogenet. 2008;186(2):115–9.
Chen SL, Qin ZY, Hu F, Wang Y, Dai YJ, Liang Y: The Role of the HOXA Gene Family in Acute Myeloid Leukemia. Genes (Basel). 2019;10(8):621.
Lawrence HJ, Sauvageau G, Humphries RK, Largman C. The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells. 1996;14(3):281–91.
Lawrence HJ, Christensen J, Fong S, Hu YL, Weissman I, Sauvageau G, Humphries RK, Largman C. Loss of expression of the Hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood. 2005;106(12):3988–94.
Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10(5):361–71.
Bhatlekar S, Fields JZ, Boman BM. HOX genes and their role in the development of human cancers. J Mol Med (Berl). 2014;92(8):811–23.
Skvarova Kramarzova K, Fiser K, Mejstrikova E, Rejlova K, Zaliova M, Fornerod M, Drabkin HA, van den Heuvel-Eibrink MM, Stary J, Trka J, et al. Homeobox gene expression in acute myeloid leukemia is linked to typical underlying molecular aberrations. J Hematol Oncol. 2014;7:94.
Ernst P, Mabon M, Davidson AJ, Zon LI, Korsmeyer SJ. An Mll-dependent Hox program drives hematopoietic progenitor expansion. Curr Biol. 2004;14(22):2063–9.
Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005;65(24):11367–74.
Dorrance AM, Liu S, Yuan W, Becknell B, Arnoczky KJ, Guimond M, Strout MP, Feng L, Nakamura T, Yu L, et al. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J Clin Invest. 2006;116(10):2707–16.
Zhang H, Zhou B, Qin S, Xu J, Harding R, Tempel W, Nayak V, Li Y, Loppnau P, Dou Y, et al. Structural and functional analysis of the DOT1L-AF10 complex reveals mechanistic insights into MLL-AF10-associated leukemogenesis. Genes Dev. 2018;32(5–6):341–6.
Andreeff M, Ruvolo V, Gadgil S, Zeng C, Coombes K, Chen W, Kornblau S, Baron AE, Drabkin HA. HOX expression patterns identify a common signature for favorable AML. Leukemia. 2008;22(11):2041–7.
Drabkin HA, Parsy C, Ferguson K, Guilhot F, Lacotte L, Roy L, Zeng C, Baron A, Hunger SP, Varella-Garcia M, et al. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia. 2002;16(2):186–95.
Buske C, Feuring-Buske M, Antonchuk J, Rosten P, Hogge DE, Eaves CJ, Humphries RK. Overexpression of HOXA10 perturbs human lymphomyelopoiesis in vitro and in vivo. Blood. 2001;97(8):2286–92.
Thorsteinsdottir U, Sauvageau G, Hough MR, Dragowska W, Lansdorp PM, Lawrence HJ, Largman C, Humphries RK. Overexpression of HOXA10 in murine hematopoietic cells perturbs both myeloid and lymphoid differentiation and leads to acute myeloid leukemia. Mol Cell Biol. 1997;17(1):495–505.
Wang H, Bei L, Shah CA, Horvath E, Eklund EA. HoxA10 influences protein ubiquitination by activating transcription of ARIH2, the gene encoding Triad1. J Biol Chem. 2011;286(19):16832–45.
Mills KI, Kohlmann A, Williams PM, Wieczorek L, Liu WM, Li R, Wei W, Bowen DT, Loeffler H, Hernandez JM, et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood. 2009;114(5):1063–72.
Bosman MC, Schepers H, Jaques J, Brouwers-Vos AZ, Quax WJ, Schuringa JJ, Vellenga E. The TAK1-NF-kappaB axis as therapeutic target for AML. Blood. 2014;124(20):3130–40.
Huang HH, Chen FY, Chou WC, Hou HA, Ko BS, Lin CT, Tang JL, Li CC, Yao M, Tsay W, et al. Long non-coding RNA HOXB-AS3 promotes myeloid cell proliferation and its higher expression is an adverse prognostic marker in patients with acute myeloid leukemia and myelodysplastic syndrome. BMC Cancer. 2019;19(1):617.
Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Bene MC, De Vos J, Hernandez JM, Hofmann WK, Mills KI, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the international microarray innovations in leukemia study group. J Clin Oncol. 2010;28(15):2529–37.
Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, Long N, Schultz AR, Traer E, Abel M, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526–31.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Dweep H, Sticht C, Gretz N. In-Silico algorithms for the screening of possible microRNA binding sites and their interactions. Curr Genomics. 2013;14(2):127–36.
Yevshin I, Sharipov R, Kolmykov S, Kondrakhin Y, Kolpakov F. GTRD: a database on gene transcription regulation-2019 update. Nucleic Acids Res. 2019;47(D1):D100–5.
Min H, Yoon S. Got target? Computational methods for microRNA target prediction and their extension. Exp Mol Med. 2010;42(4):233–44.
Paraskevopoulou MD, Vlachos IS, Karagkouni D, Georgakilas G, Kanellos I, Vergoulis T, Zagganas K, Tsanakas P, Floros E, Dalamagas T, et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44(D1):D231–8.
Tibshirani R. The lasso method for variable selection in the cox model. Stat Med. 1997;16(4):385–95.
Modhukur V, Iljasenko T, Metsalu T, Lokk K, Laisk-Podar T, Vilo J. MethSurv: a web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics. 2018;10(3):277–88.
Zhang HY, Li JH, Li G, Wang SR. Activation of ARK5/miR-1181/HOXA10 axis promotes epithelial-mesenchymal transition in ovarian cancer. Oncol Rep. 2015;34(3):1193–202.
Sun S, Su C, Zhu Y, Li H, Liu N, Xu T, Sun C, Lv Y. MicroRNA-544a regulates migration and invasion in colorectal Cancer cells via regulation of Homeobox A10. Dig Dis Sci. 2016;61(9):2535–44.
Xiao ZD, Jiao CY, Huang HT, He LJ, Zhao JJ, Lu ZY. Liu LX: miR-218 modulate hepatocellular carcinoma cell proliferation through PTEN/AKT/PI3K pathway and HoxA10. Int J Clin Exp Pathol. 2014;7(7):4039–44.
Johansen S, Brenner AK, Bartaula-Brevik S, Reikvam H, Bruserud O: The Possible Importance of beta3 Integrins for Leukemogenesis and Chemoresistance in Acute Myeloid Leukemia. Int J Mol Sci 2018, 19(1).
Wang H, Lindsey S, Konieczna I, Bei L, Horvath E, Huang W, Saberwal G, Eklund EA. Constitutively active SHP2 cooperates with HoxA10 overexpression to induce acute myeloid leukemia. J Biol Chem. 2009;284(4):2549–67.
Shah CA, Bei L, Wang H, Platanias LC, Eklund EA. HoxA10 protein regulates transcription of gene encoding fibroblast growth factor 2 (FGF2) in myeloid cells. J Biol Chem. 2012;287(22):18230–48.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Shiba N, Yoshida K, Shiraishi Y, Okuno Y, Yamato G, Hara Y, Nagata Y, Chiba K, Tanaka H, Terui K, et al. Whole-exome sequencing reveals the spectrum of gene mutations and the clonal evolution patterns in paediatric acute myeloid leukaemia. Br J Haematol. 2016;175(3):476–89.
Frohling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23(26):6285–95.
Braun BS, Shannon K. Targeting Ras in myeloid leukemias. Clin Cancer Res. 2008;14(8):2249–52.
Ney GM, Anderson B, Bender J, Kumar-Sinha C, Wu YM, Vats P, Cieslik M, Robinson DR, Li Q, Chinnaiyan AM, et al. Mutations predictive of hyperactive Ras signaling correlate with inferior survival across high-risk pediatric acute leukemia. Transl Pediatr. 2020;9(1):43–50.
Fujiwara Y, Yamaguchi H, Yui S, Tokura T, Inai K, Onai D, Omori I, Marumo A, Yamanaka S, Sakaguchi M, et al. Importance of prognostic stratification via gene mutation analysis in elderly patients with acute myelogenous leukemia. Int J Lab Hematol. 2019;41(4):461–71.
Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Dohner K, Marcucci G, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–64.
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752–9.
Pardanani A, D'Souza A, Knudson RA, Hanson CA, Ketterling RP, Tefferi A. Long-term follow-up of FIP1L1-PDGFRA-mutated patients with eosinophilia: survival and clinical outcome. Leukemia. 2012;26(11):2439–41.
Lierman E, Michaux L, Beullens E, Pierre P, Marynen P, Cools J, Vandenberghe P. FIP1L1-PDGFRalpha D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRalpha T674I eosinophilic leukemia with single agent sorafenib. Leukemia. 2009;23(5):845–51.
Golos A, Jesionek-Kupnicka D, Gil L, Braun M, Komarnicki M, Robak T, Wierzbowska A. The expression of the SLIT-ROBO family in adult patients with acute myeloid leukemia. Arch Immunol Ther Exp. 2019;67(2):109–23.
Park S, Chapuis N, Tamburini J, Bardet V, Cornillet-Lefebvre P, Willems L, Green A, Mayeux P, Lacombe C, Bouscary D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica. 2010;95(5):819–28.
Prijic S, Ugrina I, Labar B, Nemet D, Batinic J, Zadro R, Ries S, Gjadrov-Kuvedzic K, Davidovic S, Batinic D. Prognostic significance of constitutive phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase phosphorylation in acute myeloid leukemia. Leuk Lymphoma. 2015;56(8):2281–8.
Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, Vanhaesebroeck B, Muller O, Pesce F, Ifrah N, et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005;106(3):1063–6.
Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003;102(3):972–80.
Tamburini J, Elie C, Bardet V, Chapuis N, Park S, Broet P, Cornillet-Lefebvre P, Lioure B, Ugo V, Blanchet O, et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood. 2007;110(3):1025–8.
Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–45.
Piazzi M, Bavelloni A, Gallo A, Faenza I, Blalock WL: Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster. Int J Mol Sci. 2019;20(11):2718.
Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev. 2006;70(4):1032–60.
Bennett RL, Blalock WL, Abtahi DM, Pan Y, Moyer SA, May WS. RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy, and viral infection. Blood. 2006;108(3):821–9.
Hsu LC, Park JM, Zhang K, Luo JL, Maeda S, Kaufman RJ, Eckmann L, Guiney DG, Karin M. The protein kinase PKR is required for macrophage apoptosis after activation of toll-like receptor 4. Nature. 2004;428(6980):341–5.
Li YJ, Zeng JM, Huang SF, Wang XZ, Zhao SQ, Bai WJ, Cao WX, Huang ZG, Feng WL. Selective leukemia cell death by activation of the double-stranded RNA-dependent protein kinase PKR. Int J Mol Med. 2011;28(2):215–22.
Liu X, Bennett RL, Cheng X, Byrne M, Reinhard MK, May WS Jr. PKR regulates proliferation, differentiation, and survival of murine hematopoietic stem/progenitor cells. Blood. 2013;121(17):3364–74.
Blalock WL, Bavelloni A, Piazzi M, Tagliavini F, Faenza I, Martelli AM, Follo MY, Cocco L. Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress. Leukemia. 2011;25(2):236–45.
Cheng X, Byrne M, Brown KD, Konopleva MY, Kornblau SM, Bennett RL, May WS. PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice. Blood. 2015;126(13):1585–94.
Gui J, Katlinski KV, Koumenis C, Diehl JA, Fuchs SY. The PKR-like endoplasmic reticulum kinase promotes the dissemination of Myc-induced leukemic cells. Mol Cancer Res. 2019;17(7):1450–8.
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O'Dwyer KM, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–41.
Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, Senegas I, Peyraud R, Peyriga L, Theodoro F, et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia. 2013;27(11):2129–38.
Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7(7):716–35.
Auberger P, Puissant A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood. 2017;129(5):547–52.
Huang R, Liao X, Li Q. Identification and validation of potential prognostic gene biomarkers for predicting survival in patients with acute myeloid leukemia. Onco Targets Ther. 2017;10:5243–54.
Ha M, Kim JY, Han ME, Kim GH, Park SY, Jeong DC, Oh SO, Kim YH. TMEM18: a novel prognostic marker in acute myeloid leukemia. Acta Haematol. 2018;140(2):71–6.
Wang M, Lindberg J, Klevebring D, Nilsson C, Lehmann S, Gronberg H, Rantalainen M. Development and validation of a novel RNA sequencing-based prognostic score for acute myeloid leukemia. J Natl Cancer Inst. 2018;110(10):1094–101.
Linden A. Measuring diagnostic and predictive accuracy in disease management: an introduction to receiver operating characteristic (ROC) analysis. J Eval Clin Pract. 2006;12(2):132–9.
Park SM, Cho H, Thornton AM, Barlowe TS, Chou T, Chhangawala S, Fairchild L, Taggart J, Chow A, Schurer A, et al. IKZF2 drives leukemia stem cell self-renewal and inhibits myeloid differentiation. Cell Stem Cell. 2019;24(1):153–65 e157.
Papp E, Hallberg D, Konecny GE, Bruhm DC, Adleff V, Noe M, Kagiampakis I, Palsgrove D, Conklin D, Kinose Y, et al. Integrated genomic, Epigenomic, and expression analyses of ovarian Cancer cell lines. Cell Rep. 2018;25(9):2617–33.
Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, Yasunaga J, Totoki Y, Chiba K, Sato-Otsubo A, Nagae G, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47(11):1304–15.
Wang XW, Wu Y, Wang D, Qin ZF. MicroRNA network analysis identifies key microRNAs and genes associated with precancerous lesions of gastric cancer. Genet Mol Res. 2014;13(4):8695–703.
Acknowledgements
Not applicable.
Funding
This study was not supported by any funding.
Author information
Authors and Affiliations
Contributions
GC, JQQ, ZCX, GM, LZL and GYY participated in the study design. GC performed most of the microarray and RNA-seq analyses. ZCX performed cluster and classification analyses. GC drafted the paper. GC, GM and GYY participated in the final preparation and revision of the paper. All authors read and approved this final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study does not involve animal and/or human tissue/individual data/ participants, there is no ethics related issues. No permissions were required to use any repository data involved in the present study.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
GSEA results associated with HOXA10 expression derived from TCGA database.
Additional file 2: Table S2.
ORA results of HOXA10 associated DEGs.
Additional file 3: Table S3.
The detail list of exonic DMRs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Guo, C., Ju, Qq., Zhang, Cx. et al. Overexpression of HOXA10 is associated with unfavorable prognosis of acute myeloid leukemia. BMC Cancer 20, 586 (2020). https://doi.org/10.1186/s12885-020-07088-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-07088-6