
Abstract
Background
The miRNA isoforms (isomiRs) have been suggested to regulate the same pathways as the canonical miRNA and play an important biological role in miRNA-mediated gene regulation. Recently, a study has demonstrated that the presence or absence of all isomiRs could efficiently discriminate amongst 32 TCGA cancer types. Besides, an effective reduction of distinguishing isomiR features for multiclass tumor discrimination must have a major impact on our understanding of the disease and treatment of cancer.
Methods
In this study, we have constructed a combination of the genetic algorithms (GA) with Random Forest (RF) algorithms to detect reliable sets of cancer-associated 5’isomiRs from TCGA isomiR expression data for multiclass tumor classification.
Results
We obtained 100 sets of the optimal predictive features, each of which comprised of 50–5’isomiRs that could effectively classify with an average sensitivity of 92% samples from 32 different tumor types. We calculated the frequency with which a 5’isomiR found in these sets as measuring its importance for tumor classification. Many highly frequent 5’isomiRs with different 5′ loci from canonical miRNAs were detected in these sets, supporting that the isomiRs play a significant role in the multiclass tumor classification. The further functional enrichment analysis showed that the target genes of the 10 most frequently appearing 5’isomiRs were involved in the activity of transcription activator and protein kinase and cell-cell adhesion.
Conclusions
The findings of the present study indicated that the 5’isomiRs might be employed for multiclass tumor classification and the suggested that GA/RF model could perform effective tumor classification by a series of largely independent optimal predictor 5′ isomiR sets.
Similar content being viewed by others


Background
The accumulation of genetic alteration drives cancer development and progression [1]. The Cancer Genome Atlas (TCGA) consortium integrated comprehensive clinicopathologic annotation data together with molecular profiles of over 11,000 human tumors across 30 different human tumor types [2]. Analyzing these large datasets can provide more exciting opportunities to better understand the tumor characteristics and discover novel and effective predictive and prognostic tumor biomarkers and therapeutic targets. While most of the previous studies on tumor classifications have focused primarily on the gene expression data, including RNA-seq and microarray data [3,4,5]. miRNAs and isoforms of human miRNAs (isomiRs) also play essential roles and may serve as potential biomarkers for tumor classification [6,7,8,9,10,11]. The isomiRs are predominantly generated from the alternative cleavage of Drosha or Dicer and 3′addition events, which produce mature miRNA different from the canonical miRNA by a few nucleotides at the 5′ or 3′ end and designated as 5’isomiR or 3’isomiR [12, 13]. Both computational and experimental analyses revealed that the isomiRs are involved in regulating distinctive target genes and could play a crucial biological role in miRNA-mediated gene regulation [14,15,16,17,18]. In a recently conducted study, the presence or absence of 7466 isomiRs could be effectively discriminated amongst 32 TCGA cancer types with 90% sensitivity [19]. Moreover, an average sensitivity of 82% was achieved by using 456 most significant isomiRs. In the present study, we aimed to evaluate the effective reduction of discriminant isomiR features for multiclass TCGA tumor discrimination classification.
The filter, wrapper and embedded methods are typically utilized for feature selection, though all of them are not good at dealing with data which contain a large number of collinear variables. [20, 21]. The isomiRs may belong to the same miRNA family, the same miRNA cluster, or some of them even have same seed region, leading to similar or related function and highly correlated expression. In the previous studies, the wrapper method could outperform embedded methods by the combined machine learning algorithm for classification [21]. The genetic algorithms combined with the machine learning algorithm, which employs a GA as the search engine for feature subset selection and the machine learning algorithm as the classification tool, was efficiently used for classification of gene expression data [3, 4]. This algorithm could identify and classify more than 90% of samples from 31 tumor types with a set of 20 genes [3]. Beside the machine learning algorithms -- such as support vector machines (SVM), sparse representation (SR), sparse representation classifier (SRC), random forest (RF), and k-Nearest Neighbors (KNN) -- has been extensively applied in cancer prognosis and prediction analysis [3, 22,23,24,25]. Nevertheless, GA has proven ability to detect the optimal classifier effectively for multiclass cancer discrimination [4, 26]. GA is based on Darwin’s theory of natural evolution, and it is typically implemented using computer simulations in which an optimization problem is specified. GA is frequently used to generate high-quality solutions to optimization problems using genetic operators: selection, crossover, and mutation [27]. In this study, we constructed a combination of the GA with random forest algorithms to detect reliable sets of cancer-associated 5’isomiRs from TCGA isomiR expression data.
Furthermore, 5’isomiR may target very different transcripts as compared with their canonical miRNAs attributed to shifting in the seed region (typically 2–7 nt of the miRNA), which is recognized to be very critical in determining miRNA target specificity [28,29,30,31]. Various 5′ isomiRs play an important role in suppression and progression of cancer [32, 33]. Using the combined GA/RF algorithms, reliable sets of candidate tumor biomarkers for multiclass tumor discrimination was detected by combining all the miRNA isoforms with same loci of 5′ end together in TCGA isomiR expression data. In this step, the miRNA isoforms with same loci of 5′ end will be left with only one in the reliable sets, which will dramatically reduce the type of isomiRs. The findings of the present study revealed that the 5’isomiRs might be utilized for effective tumor classification and classifier can achieve an average sensitivity of 91.5% with only 50–5’isomiRs.
Methods
Datasets
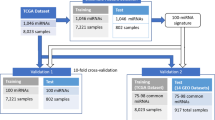
All the TCGA isomiR expression data for 10,999 TCGA datasets representing 33 tumor types was downloaded (April 2018) from the TCGA data portal (https://tcga-data.nci.nih.gov). Only primary solid tumor samples with infix ‘01’ in the TCGA sample barcode, with the exception of the blood samples derived from acute myeloid leukemia (LAML; sample infix ‘03’) were included in the study. After excluding all samples that were annotated as ‘potentially problematic’ datasets (file_annotations.txt files), 9085 eligible datasets (isoform.quantification.txt files) were finally included for further analysis (Table 1). For each dataset, we generated 5’isomiR profiles by combing all the sequences together with same loci at 5′ end, and the expression level of each 5’isomiR was calculated by the sum of all corresponding RPM (read per million) values. In order to avoid noise generated due to poorly expressed isomiRs, we only included the 5’isomiR profiles with read depths of ≥10 in more than 10 samples. Next, we log2-transformed the combined normalized read depths for each 5’isomiR; however, as the read depths ≤1 RPM were considered noise, we filtered them by assigning all values less than 1 the value 1 before log transformation.
Multiclass GA/RF classifier
In this study, we used the GA/RF based model for tumor classification. GA/RF utilizes a GA to select a set of salient features from input and classification module using RF [27]. The selected features were used as inputs to RF [22]. In a genetic algorithm, a population of strings (designated as chromosomes), which encode candidate solutions (the 5’isomiR signature in this case) to an optimization problem, evolves toward better solutions. The evolution typically starts from a population of randomly generated 5’isomiR sets and occurs in generations. In the present study, the parameters including the “chromosome” length, the “population” size and the maximum number of “generations” were set to 50 (including 50–5’isomiR set), 50, and 300, respectively. For RF classification, the randomForest was used (https://cran.r-project.org/web/packages/randomForest/randomForest.pdf). Besides, we use SVM classification for comparison, and the e1071 package in R with linear kernel function was run (https://cran.r-project.org/web/packages/e1071/index.html).
For tumor, classification prediction may vary based on different samples assigned to the training set. We repeated the above the GA/RF procedure100 times. During each of the 100 runs, the training and testing were carried out, each time using one distinct subset of randomly selected for training and the remaining subsets for testing. In a given run, the training sets were generated by randomly selecting 75% of each cancer’s available tumor datasets, and the testing sets were generated by the remaining 25% datasets. Finally, we achieved optimal 5’isomiR sets after 300 generations of GA/RF steps.
5’isomiR target prediction and function enrichment analysis
Using the TargetScanHuman (http://www.targetscan.org) and the TargetScanHuman Custom (http://www.targetscan.org/vert_50/seedmatch.html) prediction of the target genes of 5’isomiRs with original seed region along with different seed region of canonical miRNA, respectively, were performed [40]. Then, the predicted target genes were submitted to the functional annotation tools of DAVID for the functional enrichment analysis [41, 42]. For functional annotation, the 3 Gene Ontology items (GOTERM_BP_FA GOTERM_CC_FAT, and GOTERM_MF_FAT) were selected with the Enrichment Thresholds or EASE set as 0.001.
Results
Tumor classification
Here, we have constructed a combination of the genetic algorithms (GA) with Random Forest (RF) algorithms to detect reliable sets of cancer-associated 5’isomiRs from TCGA isomiR expression data for multiclass tumor classification (Fig. 1). After 100 independent runs, the prediction accuracies of each classifier for each cancer could be obtained with 300 generations of GA. Based on the initial pre-selected set population size, we obtained 100 sets of the optimal predictive features, each of which is comprised of 50–5’isomiRs. The GA/RF and GA/SVM achieved quite similar results (the average sensitivities were 92 and 91.5%, respectively), and then our following analysis only used the result from GA/RF classifier. The 100 generated predictor sets required relatively similar classification accuracies(Fig. 2a, Fig. 2b), which indicated that our selected 5’isomiR sets were remarkably accurate for multiclass tumor classifications. Besides, the prediction accuracies for cholangiocarcinoma (CHOL), rectum adenocarcinoma (READ) and esophageal carcinoma (ESCA), were recorded to be relatively low, indicating that these tumors were often classified as other types (Fig. 2c). Interestingly, the samples of these cancers could be effectively classified in some runs by altering the training and test set, with different isomiR sets, except for READ. Further, in order to investigate which tumor types could be hardly distinguished from all others, we calculated the mean prediction sensitivity for all runs. Notably, similar tumor classification was obtained as reported previously (Fig. 2d). Moreover, the majority of samples from READ tumor were misclassified as colon adenocarcinoma (COAD), which could be attributed to similar molecular expression, histology, and anatomical location [19, 34]. These findings suggested that the GA/RF model could perform effective tumor classification by a series of largely independent optimal predictor 5′ isomiR sets.

The work flow of our GA/RF based algorithm for detecting reliable sets of cancer-associated 5’isomiRs from TCGA isomiR expression data

Analysis of GA/SVM-derived optimal feature sets for 100 runs generated by GA/SVM. a The average sensitivity for 100 generated predictor sets. b The average MCC (Matthew’s Correlation Coefficient) for 100 generated predictor sets [43]. c The prediction accuracies for 32 tumor classifications. d The average sensitivity of test-set samples predicted to be each of the 32 tumor types. X-axis and Y-axis list the actual and the predicted cancer type, respectively. The color of each cell in the heatmap is the average sensitivity of the test-set samples originally as the cancer type in X-axis to be predicted as the cancer type in Y-axis
Characterization of the highly frequent 5’isomiRs
For all 100 runs, we acquired 100 sets of GA/RF classifiers and each set comprised of 50 items. Then, the frequency of selected 5′ isomiR across all the optimal predictive set was calculated. The relative significance of each 5′ isomiR for tumor classification was assessed by counting how often it appeared in these predicted optimal feature sets. For the functional enrichment analysis, we included all 5′ isomiRs with the frequency of ≥11 in these sets. It is difficult to randomly select a gene for more than 11 times in a 100 subset from 100 runs for a given dataset of 2231 samples, and the significance of adjusted p-value was calculated as less than 0.01 followed by Bonferroni correlation for multiple testing. Finally, 41 highly frequent 5’isomiRs were selected (Table 2). Notably, of which 28 had different 5′ loci than that of the canonical miRNA from miRBase 21, and some 5’isomiRs even originated from the same pre-miRNA.
Next, we examined the expression level of these highly frequent isomiRs (Fig. 3). Many of them showed higher than 10 rpm, which was the threshold value as derived from a previous study for presence or absence of isomiRs [19]. Noticeably, some of them showed extremely lower expression, nearly all of 5’isomiRs showed less than 10 rpm. It is worth noting that we combined 5′ isomiRs with same 5′ loci together in this study.

The expression level of 41 highly frequent isomiRs in 100 generated predictor sets. X-axis lists the 5’isomiR ID used in this study (Detail description can be found in Table 2). Y-axis is the log2-transformed RPM value. The line indicates the value of log2-transformed 10 rpm. The outliers are hidden
Tumor classification and functional analysis with the 9 most frequently appearing 5′ isomiRs
Next, the 9 most frequently appearing 5’isomiRs from all 100 runs were selected to determine whether a rational classification could be obtained using a reduced set. The training set and the corresponding test set were randomly produced 1000 times. Then, the RF algorithm was performed to determine the prediction accuracy of multiclass tumor classification (Fig. 4a). We could still achieve a reasonable prediction accuracy (the average sensitivity was 73.7%). In the list of 9 most frequently appearing 5’isomiRs, two 5’isomiRs were from hsa-miR-10b-5p and three 5’isomiRs belonged to hsa-miR-375. In order to investigate whether these 5’isomiRs with different 5′ loci could provide an additional contribution to the tumor classification, we removed the three 5’isomiRs which had different 5′ loci from the canonical miRNA, and randomly rebuilt the training set and the corresponding test set 1000 times again. Finally, we could only obtain an average sensitivity of 72.3% by the RF algorithm, which suggested that the isomiR shown different contribution in multiclass tumor classification than the canonical miRNA.

Tumor classification and functional enrichment analysis with the 9 most frequently appearing 5’isomiRs. a Tumor classification. Y-axis is the average sensitivity for 1000 randomly produced test sets. “a” is the 9 most frequently appearing 5’isomiRs. In the list of 9 most frequently appearing 5’isomiRs, two 5’isomiRs were from hsa-miR-10b-5p and three 5’isomiRs belonged to has-375. “b” is obtained from “a” by removing three 5’isomiRs which had different 5′ loci from the canonical miRNA (one was from hsa-miR-10b-5p and two belonged to has-375). b Bar plot shows the enriched GO terms from DAVID functional annotation analysis. The clusters integrated with enrichment score are shown as Y-axis. The –log10(P-value after correlation) is plotted on the X-axis
Furthermore, to investigate the candidate target genes of the 9 most frequently appearing 5’isomiRs, the TargetScanHuman was performed for the 5–5’isomiRs with canonical seed region and the TargetScan Custom was used for other 4–5’isomiRs with a shift in seed regions. The target genes predicted were subjected to the functional enrichment analysis. Finally, 2345 genes were recognized by the DAVID web tools to analyze the functional annotation and the detection of enriched functional categories. The gene ontology enrichment analysis suggested that these target genes were highly enriched in genes implicated in the activity of transcription activator and protein kinase and cell-cell adhesion (Fig. 4b).
Discussion
We report a novel GA/RF analytical model for multiclass tumor classification using the miRNA expression data that may reveal effective predictive and prognostic biomarkers and therapeutic targets for drug development. With an average sensitivity of 92%, we were able to accurately classify the tumor samples using 100 different 50–5’isomiR sets, though some 5’isomiRs appeared repetitively in the sets. These predictive 5’isomiRs sets could achieve similar prediction accuracies with slight overlap; suggesting that even less sensitive 5’isomiRs could be detected for the tumor classification. Notably, most of the tumor types could be easily distinguished with high sensitivity. However, there were also some cancers that exhibited low prediction accuracy due to similar histology and anatomical location [19, 34]. In this study, we used GA algorithm to obtain the optimal isomiR set for maximizing the prediction accuracy in all TCGA cancer types but not for some individual cancer. Therefore, the samples from some cancers that cannot be classified by one set may be successfully classified by another set. In addition, we calculated the frequency with which each 5’isomiRs appeared in these sets. More than half of 41 highly frequent 5′ isomiRs showed different 5′ loci than the canonical miRNA, supporting that the isomiRs play a significant role in the multiclass tumor classification. It is noted that our analysis only included tumor samples, and we cannot distinguish cancer-specific isomiRs from tissue-specific biomarkers. Actually, a group from Saarland university had utilized a tissue specificity index to define the distribution of miRNA across 61 tissue biopsies of two individuals, and people can check whether the detected isomiRs correspond to the tissue-specific miRNA expression in their web-based repository [35].
In a recent study, the RNA-seq expression data analysis revealed that many development-related genes are essential for the analysis of TCGA cancer classification [3]. Similar clues were also revealed in the present study. 5’isomiR-233, one of the most frequently appearing 5’isomiRs in 100 generated predictor sets, derived from the shift in the seed region of canonical hsa-mir-196b-5p, which usually appears to be expressed from the intragenic regions of HOX gene clusters that are major regulators of animal development [36]. Increasing studies have suggested that 5’isomiR-313, combined from the isomiRs with identical 5′ loci of the canonical hsa-miR-205-5p, play an important role in normal cellular development as well as in cancer development [37, 38]. Moreover, TBX5, one of the most important genes for tumor classification from the previous study [3], could be regulated by one of the 5 most frequently appearing 5’isomiRs in our sets (miR-10b-5p/5’isomiR-39) as derived from the TargetScanHuman prediction.
Using only 50–5’isomiRs, the present GA/RF model could achieve comparable prediction performances consistent with previous report, with an average accuracy of 90% for all isomiRs [19]. We also detected the similar discriminatory isomiRs as their finding. For example, the isomiRs of has-miR-205-5p and has-miR-944, two of the most important miRNAs detected by the method using the presence or absence of isomiRs amongst 32 TCGA cancer types, are also listed in the ten highly frequent isomiRs from 100 generated predictor sets. The isomiR of hsa-mir-196b-5p, the most frequently appearing 5’isomiRs with a shift in seed regions found in our study, showed a high VI score in previous report [19]. Further, we reduced the number of features by employing two strategies. In the first approach, we combined the isomiR with same 5′ loci to reduce the type of isomiRs. While in the second approach, the GA-based isomiR selection reduced the feature selection significantly. We also found that the 9 most frequently appearing 5’isomiRs could achieve an average sensitivity of 73.7%, suggesting that a reasonable accurate performance could be obtained with less number of features. The features can be further reduced by additional approaches, including hybrid GA-based machine learning method [39]. The highly expressed 5’isomiRs (rpm > 10 in all samples) and slightly expressed 5’isomiRs (rpm < 10 nearly in all samples), demonstrated that the expression level of isomiRs could also be beneficial for the tumor classification.
Conclusions
In conclusion, the present study demonstrated that the 5’isomiRs might be employed for multiclass tumor classification and the suggested that GA/RF model could perform effective tumor classification by a series of largely independent optimal predictor 5′ isomiR sets.
Abbreviations
- AML:
-
Acute myeloid leukemia
- CHOL:
-
Cholangiocarcinoma
- COAD:
-
Colon adenocarcinoma
- ESCA:
-
Esophageal carcinoma
- GA:
-
Genetic algorithms
- isomiR:
-
MiRNA isoform
- MCC:
-
Matthew’s Correlation Coefficient
- READ:
-
Rectum adenocarcinoma
- RF:
-
Random forest
- RPM:
-
Read per million
- SVM:
-
Support vector machine
- TCGA:
-
The Cancer Genome Atlas
References
Herceg Z, Hainaut P. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Mol Oncol. 2007;1:26–41.
Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, KRM S, Ozenberger BA, et al. The Cancer genome atlas Pan-Cancer analysis project. Nat genet. Nat Publ Group. 2013;45:1113–20.
Li Y, Kang K, Krahn JM, Croutwater N, Lee K, Umbach DM, et al. A comprehensive genomic pan-cancer classification using the Cancer genome atlas gene expression data. BMC genomics. BioMed Central. 2017;18:508.
Liu JJ, Cutler G, Li W, Pan Z, Peng S, Hoey T, et al. Multiclass cancer classification and biomarker discovery using GA-based algorithms. Bioinformatics. 2005;21:2691–7.
Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378:1812–23.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer; Nature Publishing Group2006;6:857–866.
Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature; Nature Publishing Group2005;435:834–838.
Pimentel F, Bonilla P, Ravishankar YG, Contag A, Gopal N, LaCour S, et al. Technology in MicroRNA profiling: circulating MicroRNAs as noninvasive Cancer biomarkers in breast Cancer. J Lab Autom. 2015;20:574–88.
Etheridge A, Lee I, Hood L, Galas D, Wang K. Extracellular microRNA: a new source of biomarkers. Mutat Res. 2011;717:85–90.
Yan W, Xu L, Sun Z, Lin Y, Zhang W, Chen J, et al. MicroRNA biomarker identification for pediatric acute myeloid leukemia based on a novel bioinformatics model. Oncotarget. Impact Journals; 2015;6:26424–26436.
Neilsen CT, Goodall GJ, Bracken CP. IsomiRs--the overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012;28:544–9.
McCall MN, Kim M-S, Adil M, Patil AH, Lu Y, Mitchell CJ, et al. Toward the human cellular microRNAome. Genome Res. 2017;27:1769–81.
Mercey O, Popa A, Cavard A, Paquet A, Chevalier B, Pons N, et al. Characterizing isomiR variants within the microRNA-34/449 family. FEBS Lett. 2017;591:693–705.
Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. 2013;14:475–88.
Hinton A, Hunter SE, Afrikanova I, Jones GA, Lopez AD, Fogel GB, et al. sRNA-seq analysis of human embryonic stem cells and definitive endoderm reveal differentially expressed microRNAs and novel isomiRs with distinct targets. Stem Cells. 2014.
Telonis AG, Loher P, Jing Y, Londin E, Rigoutsos I. Beyond the one-locus-one-miRNA paradigm: microRNA isoforms enable deeper insights into breast cancer heterogeneity. Nucleic Acids Res. 2015;43:9158–75.
Wang S, Tu J, Wang L, Lu Z. Entropy-Based model for MiRNA isoform analysis. PLoS One. 2015;10:e0118856.
Telonis AG, Magee R, Loher P, Chervoneva I, Londin E, Rigoutsos I. Knowledge about the presence or absence of miRNA isoforms (isomiRs) can successfully discriminate amongst 32 TCGA cancer types. Nucleic Acids Res. 2017;45:2973–85.
Kang J, Rancati T, Lee S, Oh JH, Kerns SL, Scott JG, et al. Machine learning and Radiogenomics: lessons learned and future directions. Front Oncol. 2018;8:228.
Rodriguez-Galiano VF, Luque-Espinar JA, Chica-Olmo M, Mendes MP. Feature selection approaches for predictive modelling of groundwater nitrate pollution: an evaluation of filters, embedded and wrapper methods. Sci Total Environ. 2018;624:661–72.
Noble WS. What is a support vector machine? Nat Biotechnol. 2006;24:1565–7.
Kourou K, Exarchos TP, Exarchos KP, Karamouzis MV, Fotiadis DI. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotechnol J. 2015;13:8–17.
Statnikov A, Wang L. Aliferis CF. a comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC bioinformatics. BioMed Central. 2008;9:319.
Gan B, Zheng C-H, Zhang J, Wang H-Q. Sparse representation for tumor classification based on feature extraction using latent low-rank representation. Biomed Res Int Hindawi. 2014;2014:420856–7.
Mukhopadhyay A, Bandyopadhyay S, Maulik U. Multi-class clustering of Cancer subtypes through SVM based Ensemble of Pareto-Optimal Solutions for gene marker identification. Navarro A, editor PLoS ONE Public Library of Science; 2010;5:e13803.
Genetic algorithms in search, optimization, and machine learning. Choice Reviews Online. 1989;27:27–0936–27–0936.
Tan GC, Chan E, Molnar A, Sarkar R, Alexieva D, Isa IM, et al. 5′ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014;42:9424–35.
Wang X. Composition of seed sequence is a major determinant of microRNA targeting patterns. Bioinformatics. 2014;30:1377–83.
Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105.
Wang S, Xu Y, Li M, Tu J, Lu Z. Dysregulation of miRNA isoform level at 5 “end in Alzheimer”s disease. Gene. 2016.
Salem O, Erdem N, Jung J, Münstermann E, Wörner A, Wilhelm H, et al. The highly expressed 5'isomiR of hsa-miR-140-3p contributes to the tumor-suppressive effects of miR-140 by reducing breast cancer proliferation and migration. BMC genomics. BioMed Central. 2016;17:566.
Magee RG, Telonis AG, Loher P, Londin E, Rigoutsos I. Profiles of miRNA isoforms and tRNA fragments in prostate Cancer. Sci Rep; Nature Publishing Group2018;8:5314.
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature; Nature Publishing Group2012;487:330–337.
Ludwig N, Leidinger P, Becker K, Backes C, Fehlmann T, Pallasch C, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865–77.
Yekta S, Shih I-H, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–6.
Gulei D, Magdo L, Jurj A, Raduly L, Cojocneanu-Petric R, Moldovan A, et al. The silent healer: miR-205-5p up-regulation inhibits epithelial to mesenchymal transition in colon cancer cells by indirectly up-regulating E-cadherin expression. Cell Death Dis; Nature Publishing Group2018;9:66.
De Cola A, Volpe S, Budani MC, Ferracin M, Lattanzio R, Turdo A, et al. miR-205-5p-mediated downregulation of ErbB/HER receptors in breast cancer stem cells results in targeted therapy resistance. Cell death dis. Nat Publ Group; 2015;6:e1823–e1823.
Huerta EB, Duval B, Hao J-KA, Hybrid GA. SVM approach for gene selection and classification of microarray data. Applications of evolutionary computing. Berlin, Heidelberg: Springer Berlin Heidelberg; 2006. p. 34–44.
Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4:101.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13.
Gorodkin J. Comparing two K-category assignments by a K-category correlation coefficient. Comput Biol Chem. 2004;28:367–74.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (61601332, 31301148), the Zhejiang Provincial Natural Science Foundation of China (LQ16F010009), the Public Welfare Technology Application Research Plan Project of Zhejiang Science and Technology Department(2017C32103) and the Special Science and Technology Innovation Project for Seeds and Seedlings of Wenzhou City(N20160017). The funders played no role in study design, data collection, analysis or interpretation of the data.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
SW and MW conceived and designed the study, SW and ZZ performed the analysis, and interpreted the results. PC interpreted the results. SW wrote the manuscript. All authors approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, S., Zheng, Z., Chen, P. et al. Tumor classification and biomarker discovery based on the 5’isomiR expression level. BMC Cancer 19, 127 (2019). https://doi.org/10.1186/s12885-019-5340-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-019-5340-y