
Abstract
Background
Deletions of 6q15–16.1 are recurrently found in pediatric T-cell acute lymphoblastic leukemia (T-ALL). This chromosomal region includes the mitogen-activated protein kinase kinase kinase 7 (MAP3K7) gene which has a crucial role in innate immune signaling and was observed to be functionally and prognostically relevant in different cancer entities. Therefore, we correlated the presence of MAP3K7 deletions with clinical parameters in a cohort of 327 pediatric T-ALL patients and investigated the function of MAP3K7 in the T-ALL cell lines CCRF-CEM, Jurkat and MOLT-4.
Methods
MAP3K7 deletions were detected by multiplex ligation-dependent probe amplification (MLPA). T-ALL cell lines were transduced with adeno-associated virus (AAV) vectors expressing anti-MAP3K7 shRNA or a non-silencing shRNA together with a GFP reporter. Transduction efficiency was measured by flow cytometry and depletion efficiency by RT-PCR and Western blots. Induction of apoptosis was measured by flow cytometry after staining with PE-conjugated Annexin V. In order to assess the contribution of NF-κB signaling to the effects of MAP3K7 depletion, cells were treated with TNF-α and cell lysates analyzed for components of the NF-κB pathway by Western blotting and for expression of the NF-κB target genes BCL2, CMYC, FAS, PTEN and TNF-α by RT-PCR.
Results
MAP3K7 is deleted in approximately 10% and point-mutated in approximately 1% of children with T-ALL. In 32 of 33 leukemias the deletion of MAP3K7 also included the adjacent CASP8AP2 gene. MAP3K7 deletions were associated with the occurrence of SIL-TAL1 fusions and a mature immunophenotype, but not with response to treatment and outcome. Depletion of MAP3K7 expression in T-ALL cell lines by shRNAs slowed down proliferation and induced apoptosis, but neither changed protein levels of components of NF-κB signaling nor NF-κB target gene expression after stimulation with TNF-α.
Conclusions
This study revealed that the recurrent deletion of MAP3K7/CASP8AP2 is associated with SIL-TAL1 fusions and a mature immunophenotype, but not with response to treatment and risk of relapse. Homozygous deletions of MAP3K7 were not observed, and efficient depletion of MAP3K7 interfered with viability of T-ALL cells, indicating that a residual expression of MAP3K7 is indispensable for T-lymphoblasts.
Similar content being viewed by others


Background
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive malignancy of thymocyte progenitors and constitutes 10–15% of pediatric ALL [1]. With current treatment protocols, approximately 80% of children suffering from T-ALL are cured [2, 3]. In a series of 73 primary T-ALL patient samples, Remke et al. (2009) identified recurrent deletions of 6q14.1–14.3 and 6q15–16.1 that were associated with an unfavorable response to treatment [4]. Of the 16 genes localized in these regions, the mRNA expression of CASP8AP2 has been shown to be most strongly affected by the deletion and CASP8AP2 has been suggested to be a tumor suppressor [4]. Similar 6q15 deletions were found in several cohorts of childhood ALL and T-cell lymphoblastic lymphomas (T-LBL) [5,6,7,8,9,10] as well as in other hematological malignancies [11,12,13,14] and in solid tumors like breast, gastric and prostate cancer [15,16,17,18] (Fig. 1). By whole exome sequencing of T-ALL samples, we had found recurrent (2/13) mutations in the MAP3K7 gene which is included within the commonly deleted region 6q15–16.1 [19].

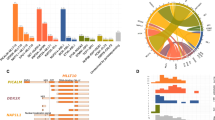
Regions of minimal deletion (RMD) on chr6q in pediatric acute lymphoblastic leukemia (ALL) and/or T-cell lymphoblastic lymphoma (T-LBL) as identified in published studies since 2004. The RMD results were derived from array comparative genomic hybridization (CGH)1, loss of heterozygosity analysis (LOH)2, single nucleotide polymorphism array analysis (SNP array)3 and fluorescence in situ hybridization (FISH)4. Originally described nucleotide positions of the proximal and distal boundaries of each RMD were adjusted to the current reference genome GRCh38/hg38 (released in 12/2013). Relative positions of previously suggested potential tumor suppressor genes (EPHA7, GRIK2) and CASP8AP2/MAP3K7 are shown by dashed lines. A RMD derived from studies of pediatric B- and/or T-ALL samples. B RMD derived from studies in T-LBL
MAP3K7 codes for a mitogen-activated protein kinase that has alternatively been termed TAK1 (transforming growth factor beta activated kinase 1) [20]. It is involved in various inflammatory and immune signaling pathways like T-cell receptor, Toll-like receptor and TNF-α signaling [21]. Several stimuli lead to association with TAK1-binding proteins (TAB1–3) [22] followed by an activation of other mitogen-activated protein kinases (ERK, JNK, p38) and the transcription factor NF-κB [23]. Consistently, MAP3K7 knockout in mice resulted in NF-κB inactivation in T-cells [24] and led to the development of myelomonocytic leukemia [25]. However, its distinct biological functions and relevance for different tumor entities appear to be cell type-specific and remain controversial [26]. For instance, in prostate cancer MAP3K7 deletion is associated with an advanced tumor stage, lymph node metastasis and an early biochemical recurrence [16, 27], and suppression of MAP3K7 has been shown to promote tumorigenesis [28]. In contrast, inhibition of MAP3K7 in breast cancer cells reduced tumor growth and impaired metastasis [29,30,31]. In an AML xenograft model, inhibition of MAP3K7 attenuated leukemia development [32].
In order to investigate the clinical relevance of deletions of MAP3K7 in pediatric T-ALL, we analyzed a cohort of 327 primary T-ALL patient samples for MAP3K7 deletions and correlated MAP3K7 status with clinical features. The functional relevance of reduced MAP3K7 expression in T-ALL cell lines was investigated by analyzing the effects of shRNA-mediated MAP3K7 depletion on cell proliferation, apoptosis and NF-κB activation.
Methods
Patients
Patients were treated according to ALL-BFM 2000 [3] or AIEOP-BFM ALL 2009 protocols. These trials were registered at www.clinicaltrials.gov (#NCT00430118 and #NCT01117441). The institutional review boards of Hannover Medical School (Nr. 2522, November 9th, 2000 and December 22nd, 2008) and University of Schleswig-Holstein (A 177/09, March 12th, 2010) approved the trials. Informed consent was obtained in accordance with the declaration of Helsinki. For further details refer to the Additional file 1: Supplemental Methods.
MLPA
Multiplex ligation-dependent probe amplification (MLPA) was performed using the MRC Holland (Amsterdam, The Netherlands) SALSA MLPA probe mix P383-A1 TALL with three additional probes for the MAP3K7 gene according to the manufacturer’s instructions. The MAP3K7 probe sequences were:
-
Exon 1: 5´ GGGTTCCCTAAGGGTTGGACATGTCTACAGCCTCTGCCGCCTCCTCCTCCTCCTCGTCTTC/ GGCCGGTGAGATGATCGAAGCCCCTTCCCAGGTCCTCAACTCTAGATTGGATCTTGCTGGCAC 3´
-
Exon 5: 5´ GGGTTCCCTAAGGGTTGGACCACGCAATGAGTTGGTGTTTACAGTG/ TTCCCAAGGAGTGGCTTATCTTCACAGCATGCAACCCAAAGCGTCTAGATTGGATCTTGCTGGCAC 3´
-
Exon 7: 5´ GGGTTCCCTAAGGGTTGGACGTCTTCAGCTGGGGTATTATTCTTTGGGAAGTGATAACGCG/ TCGGAAACCCTTTGATGAGATTGGTGGCCCAGCTTCTAGATTGGATCTTGCTGGCAC 3´
Polymerase chain reaction (PCR) products were separated by capillary electrophoresis on an ABI-3130XL device. The size standard was GeneScan 500–250 (both Applied Biosystems).
Cell culture
HEK293T cells were cultured in DMEM medium and CCRF-CEM, MOLT-4 and Jurkat cells in RPMI 1640 medium (both Gibco). Media were supplemented with 10% fetal bovine serum and 100 μg/mL penicillin-streptomycin (Biochrom). All cell lines were cultured at 37 °C and 5% CO2.
AAV vector production
AAV vectors were produced as described previously [33, 34]. Briefly, HEK293T cells were triple-transfected with equal amounts of an AAV helper plasmid (encoding AAV rep and cap genes), an AAV vector plasmid encoding the anti-MAP3K7 shRNA or a non-silencing shRNA and an adenoviral helper construct. The cap gene AAVrh10A2 was used for the CCRF-CEM cell line and AAV9A2 for the Jurkat and MOLT-4 cell lines. These are synthetic AAV cap genes that were created through insertion of short re-targeting peptides into wild-type AAVrh10 or AAV9, as recently described [35]. Sense strand nucleotide sequences for the three anti-MAP3K7 shRNAs were (positions according to hg38):
-
shRNA 1 5’ GTGTGTCTTGTGATGGAATA 3′, chr 6:90,561,667–90,561,622
-
shRNA 2 5’ GCAAGTTCCTGCCACAAATGA 3′, chr 6:90,548,099–90,548,119
-
shRNA 3 5’ GGACATTGCTTCTACAAATAC 3′, chr 6:90,548,147–90,548,167.
Sense strand nucleotide sequence of non-silencing control shRNA:
-
shRNA ns 5’ GTAACGACGCGACGACGTAA 3’
See Additional file 1: Supplemental Methods for further details on shRNA design and cloning, as well as on AAV vector production.
Transduction of T-ALL cell lines
T-ALL cells were seeded at a density of 40 cells/μl in 12-well plates and transduced with the shRNA-encoding AAV vectors at a multiplicity of infection (MOI, i.e., vector genomes per cell) between 1*104 to 5*105. Cells were incubated for 72 h, spun down and resuspended in fresh medium. For further experiments, cells were either re-seeded at a density of 40 cells/μl for proliferation analysis or fixed by adding paraformaldehyde (PFA) to a final concentration of 4% in phosphate-buffered saline (PBS, Sigma) and incubating for 30 min at room temperature. After washing with PBS, transduction rates were measured by detection of the green fluorescent protein (GFP) reporter that is co-encoded by all AAV/shRNA vectors by means of flow cytometry (Cytomics FC500 MPL analyzer, Beckman Coulter).
Annexin V cell death assay
Six days after transduction, T-ALL cells were counted and pelleted for staining with an Annexin V-R-Phycoerythrin (PE) conjugate (BD Biosciences). Cells were washed twice in Annexin V binding buffer and then incubated with Annexin V-PE for 15 min at room temperature. Next, they were fixed with 4% PFA/PBS, washed once with PBS, and analyzed by flow cytometry. See Additional file 1: Figure S3 for FACS dot plots and Additional file 1: Supplemental Methods for further description.
Quantitative real-time-PCR (qRT-PCR)
Six days after transduction, total RNA was extracted by using the RNeasy Mini kit (Qiagen) in accordance with the manufacturer’s instructions. cDNA was synthesized by using oligo(dT) primers and RevertAid H Minus M-MuLV Reverse Transcriptase (Thermo Scientific) according to the manufacturer’s instructions. All quantitative RT-PCR reactions were performed in triplicates by an ABI StepOnePlus thermocycler (Applied Biosystems) with SYBR Green PCR Master Mix (Thermo Scientific). Primer sequences are listed in Additional file 1: Table S1.
Western blotting
To obtain whole cell lysates, cells were extracted six days after transduction by use of Mammalian Protein Extraction Reagent (M-PER, Thermo Fisher Scientific) and subjected to at least three freeze-thaw cycles (− 80 °C/room temperature). Samples were run on a sodium-dodecyl sulfate/ 10% polyacrylamide gel and blotted onto a Westran S polyvinylidene difluoride membrane (GE Health Care). After blocking for 24 h in a 5% milk solution, proteins of interest were detected by incubation for 24 h with antibodies against MAP3K7, NF-κB p100/p52, NF-κB p105/p50, IκB (all Cell Signaling) used at a 1:1000 dilution in 5% milk/TBS-T (Tris-buffered saline with 0.5% Tween-20) or 5% BSA (bovine serum albumin)/TBS-T for phospho NF-κB p65 Ser536 (Cell Signaling). Incubation for 1 h with a peroxidase-conjugated anti-mouse or anti-rabbit antibody (both Sigma) was followed by signal detection with Western Lightning Plus-ECL detection reagent (PerkinElmer) using the Fusion FX detection system (Vilber Lourmat). ImageJ (1.48v) was used for quantification of results.
Statistical analysis and graph preparation
Event-free survival (EFS) was defined as the time from diagnosis to the date of last follow-up in complete remission or first event. Events were non-response (defined as not achieving a complete remission after the first HR-block in ALL-BFM 2000 and after the third HR-block in AIEOP-BFM ALL 2009), relapse, secondary neoplasm, or death from any cause. Failure to achieve remission due to early death or non-response was considered as event at time zero. Survival was defined as the time of diagnosis to death from any cause or last follow-up. The Kaplan-Meier method was used to estimate survival rates, and differences were compared with the two-sided log rank test. Cox’s proportional hazards model was used for uni- and multivariate analyses. Cumulative incidence (CI) functions for competing events were constructed by the method of Kalbfleisch and Prentice, and were compared with the Gray’s test [36]. Results are presented as estimated probability of 5-year EFS (pEFS) and estimated cumulative incidence of relapse (pCIR) with standard error (± SE). Differences in the distribution of individual parameters among patient subsets were analyzed using Fisher’s exact test for categorized variables and the Mann-Whitney-U test for continuous variables. Logistical regression was used to analyze the effect of mutations on response variables (prednisone response, MRD). All statistical analyses were conducted using the SAS program (SAS-PC, v. 9.1, SAS Institute Inc.).
Experimental results were analyzed by Student’s t-tests as well as one-way and two-way ANOVA. If not stated otherwise in the figure legends, data are presented as average ± SE. Graphs and tables were prepared by using GraphPad Prism6, Microsoft Office 2010 (Excel, PowerPoint) and ImageJ 1.48v. Coffalyser software was used for MLPA analyses (available at http://www.mlpa.com).
Results
Deletions of MAP3K7 are associated with SIL-TAL1 fusions and with a mature T-ALL immunophenotype
We performed MLPA on 327 primary T-ALL patient samples to detect deletions of 6q15 using probes directed against MAP3K7 and CASP8AP2. All patients had been treated in the ALL-BFM 2000 or in the AIEOP-BFM ALL 2009 studies and clinical data were available (Table 1). All deletions of 6q15 were heterozygous. Thirty-two of 33 samples with a MAP3K7 deletion also showed a loss of the adjacent CASP8AP2 gene. We assume that all three genes located between MAP3K7 and CASP8AP2 (GJA10, BACH2, MIR4464) and a variable number of adjacent genes are co-deleted in these leukemias. Only in one sample a MAP3K7 deletion without a CASP8AP2 deletion was detected. These results support earlier results [4,5,6,7,8,9] showing that 6q15 deletions usually affect several genes. Analysis of clinical data of all 327 pediatric T-ALL patients who had been uniformly treated with ALL-BFM protocols revealed that the MAP3K7/CASP8AP2 deletion is significantly associated with a mature T-ALL immunophenotype (p = 0.0005; Table 1), but not with any other clinical feature. Specifically, response to treatment as measured by prednisone response on day 8 and MRD assessment on days 33 and 78 after start of induction treatment did not differ between patients with or without a MAP3K7 deletion (Table 1). There was no association of MAP3K7/CASP8AP2 deletions with the cumulative incidence of relapse or overall survival (Fig. 2a). MAP3K7/CASP8AP2 deletions were significantly more frequently observed in SIL-TAL1 positive than in SIL-TAL1 negative T-ALLs (p = 0.005). No other genetic feature including NOTCH1 and PTEN mutations were associated with MAP3K7 deletion. There was a trend for SIL-TAL1 positive T-ALL patients harboring a MAP3K7/CASP8AP2 deletion towards a higher risk of relapse compared to patients with SIL-TAL1 fusion, but without MAP3K7/CASP8AP2 deletion (p(Gray) = 0.13; Fig. 2b). Targeted sequencing identified a monoallelic point mutation in MAP3K7 in less than 1% (1 of 147) of a subgroup of the T-ALL patients [37].

Deletion of MAP3K7/CASP8AP2 does not affect outcome of pediatric T-ALL patients. Cumulative incidence of relapse (pCIR) and probability of event-free survival (pEFS) for T-ALL patients enrolled in ALL-BFM 2000 and ALL-BFM 2009 with MAP3K7/CASP8AP2 wild type (blue) or MAP3K7/CASP8AP2 deletion (red). Results are presented as estimated probability of 5-year EFS (pEFS) and estimated cumulative incidence of relapse (pCIR) with standard error (± SE). a T-ALL patients with or without MAP3K7/CASP8AP2 deletion (n = 327). MAP3K7/CASP8AP2 deletion neither affects the pEFS (p(Log-Rank) = 0.4) nor pCIR (p(Gray) = 0.98). b T-ALL patients with SIL-TAL1 fusion (n = 52) who do or do not carry an additional MAP3K7/CASP8AP2 deletion. SIL-TAL1 positive patients harboring a deletion of MAP3K7/CASP8AP2 show a trend towards a higher pCIR (p(Gray) = 0.13)
T-ALL cell lines can be efficiently transduced by adeno-associated viral vectors
To analyze the biological effects of a reduced MAP3K7 expression, we chose three T-ALL cell lines (CCRF-CEM, Jurkat, MOLT-4) that do not carry a MAP3K7 deletion as assessed by MLPA. MAP3K7 mRNA expression levels were estimated by quantitative RT-PCR to be similar in all three cell lines and comparable to those in HEK293 cells (Additional file 1: Figure S1 A). We aimed at phenocopying a MAP3K7 deletion by shRNA-mediated depletion. Because T-ALL cell lines are resistant to common methods of chemical transfection [38], we used an adeno-associated viral (AAV) vector-mediated transduction system to efficiently deliver shRNA to our cell lines. Flow cytometry analysis demonstrated transduction efficiencies mostly above 80% for the cell lines CCRF-CEM and Jurkat (Additional file 1: Figure S1 B). All subsequent experiments with these cell lines were performed after a transduction efficiency of at least 80% had been confirmed. However, transduction of MOLT-4 cells was less efficient and depended on the type of shRNA, so the threshold for further analysis in this cell line was set to a transduction efficiency of at least 60%. Depletion efficiency as assessed by quantitative RT-PCR and Western blotting was uniform, with both MAP3K7 mRNA and MAP3K7 protein being in the range of 25% of that after treatment with non-silencing shRNA (Additional file 1: Figure S1C-D).
MAP3K7 knockdown slows down proliferation in T-ALL cell lines
Treatment with anti-MAP3K7 shRNA significantly slowed down proliferation in all three of the T-ALL cell lines analyzed (Fig. 3a). Five days after transduction, all three shRNAs resulted in a significant reduction of cell numbers compared to the non-silencing control (two-way ANOVA for treatment vs non-silencing control: p(CCRF-CEM) = 0.0044, p(Jurkat) = 0.0057, p(MOLT-4) = < 0.0001). Treatment with shRNA 1 in Jurkat and MOLT-4 cells completely abrogated cell proliferation and/or led to a loss of cells.

MAP3K7 depletion decreases proliferation and induces apoptosis in T-ALL cell lines. T-ALL cell lines were transduced with AAV vectors coding for three different shRNAs (1, 2, 3) and one non-silencing shRNA (ns). Transduction efficiency was controlled by flow cytometry after 72 h of incubation. a MAP3K7 depletion reduces proliferation of T-ALL cells. After exchanging the culture medium three days after transduction, cells were seeded at a density of 40 cells/μl. Every 24 h, an aliquot was stained by Trypan blue and vital cells were counted in a hemocytometer (Neubauer improved, Assistent). Relative proliferation was defined as the ratio of cell numbers on the day of interest over the starting cell number. Means of relative proliferation and standard deviations are given in the graph. Significance of differences in proliferation rates were calculated by two-way ANOVA and Student’s t-test compared to shRNA ns on day 5 of counting (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** p < 0.0001, n(CCRF-CEM, MOLT-4) = 3, n(Jurkat) = 5). b MAP3K7 depletion sensitizes T-ALL cells for apoptosis. Six days after transduction, apoptotic cells were stained with PE-conjugated Annexin V and measured by flow cytometry. Dot plot gates for untreated control were set to have less than 1% apoptotic cells. Gates of transduced cells were adjusted accordingly. The percentage of Annexin V-positive cells was compared between treatment with non-silencing shRNA (ns) and shRNAs 1, 2 and 3 directed against MAP3K7. Results are presented as means and standard deviations of PE-positive cells in percent. Significance was calculated by unpaired t-test compared to non-silencing shRNA (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** p < 0.0001, n(CCRF-CEM, Jurkat) = 3, n(MOLT-4) = 6)
MAP3K7 depletion induces apoptosis in T-ALL cell lines
In order to assess if decreasing cell numbers after depletion of MAP3K7 were due to a higher rate of apoptosis, we performed an Annexin V assay six days after transduction. Treatment with anti-MAP3K7 shRNA resulted in an increase in the proportion of Annexin V positive cells in all three cell lines of up to 8-fold (Fig. 3b). The extent of this effect depended on the cell type and the type of shRNA used. In all three cell lines, shRNA 2 had no or only a marginal effect on the proportion of Annexin V positive cells, possibly reflecting the relative low transduction efficiency for the corresponding shRNA construct. The strongest reduction of cell numbers was observed with the shRNA that most strongly induced apoptosis, suggesting that depletion of MAP3K7 impairs cell density by increasing the rate of apoptosis. However, the potential to induce apoptosis did not fully explain the effect on cell numbers, indicating that further mechanisms influence proliferation.
Effects of MAP3K7 depletion are not mediated by NF-κB inactivation
MAP3K7 deficiency has been shown to inhibit NF-κB activation in different cell types [29, 32, 39]. To study the relevance of this interaction in T-ALL cells, we stimulated cell lines with TNF-α six days after transduction with shRNAs against MAP3K7 and analyzed protein lysates by Western blotting for components of the NF-κB pathway (Fig. 4). Stimulation with TNF-α led to rapid and transient degradation of IκB and accumulation of phospho-NF-κB p65 (Ser536) in both untreated control cells and ns shRNA-treated cells, while other NF-κB proteins (p100, p105, p50) remained largely unaffected (Fig. 4a). Depletion of MAP3K7 mediated by shRNAs 1 and 3 neither changed IκB levels in unstimulated cells nor did it prevent the degradation of IκB after stimulation with TNF-α. shRNA 2 partially impaired degradation of IκB (Fig. 4b).

MAP3K7 depletion does not inhibit NF-κB activation after stimulation of T-ALL cell lines with TNF-α. Six days after transduction with anti-MAP3K7 shRNA, T-ALL cell lines were stimulated with TNF-α (10 ng/μl) for 30 min and whole cell lysates were analyzed by Western blotting. a Stimulation of non-transduced T-ALL cells results in degradation of IκB. b MAP3K7 depletion does not prevent degradation of IκB after stimulation with TNF-α. c Treatment with anti-MAP3K7 shRNA did not consistently change expression patterns of NF-κB target genes in T-ALL cell lines. Six days after transduction with anti-MAP3K7 shRNA, total RNA was extracted, cDNA was synthesized and MAP3K7 mRNA expression quantified by qRT-PCR. HPRT1 was used as internal control. Mean values and SE of expression levels are given (n = 3). CCRF-CEM carries a PTEN deletion and does not express PTEN [62]
Next, we asked if MAP3K7 depletion influences expression levels of NF-κB target genes. We extracted total RNA from T-ALL cells after MAP3K7 depletion and analyzed mRNA expression levels of five NF-κB target genes (BCL2 [40], CMYC [41], PTEN [42], TNF-α [43, 44], FAS [45, 46]) by quantitative RT-PCR. Expression of none of these genes was consistently changed by more than two-fold in any of the cell lines (Fig. 4c), indicating that the effect of NF-κB on gene expression does not depend on MAP3K7. We conclude that in T-ALL cell lines, MAP3K7 is not required for the degradation of IκB and subsequent activation of NF-κB after stimulation with TNF-α.
Discussion
We confirmed in a large cohort that heterozygous deletions of 6p15, including the MAP3K7 locus, are a frequent event in T-ALL. In contrast, point mutations in MAP3K7 were observed in less than 1% of primary T-ALLs [37]. MAP3K7 deletions were associated with the presence of SIL-TAL1 fusions and a mature immunophenotype. Although leukemias with TAL1 overexpression were described to reflect the late cortical stage of thymocyte differentiation [47], SIL-TAL1 fusions were not associated with a certain immunophenotype in our and published cohorts [48]. We therefore hypothesize that the deletion of MAP3K7/CASP8AP2 or another gene on 6q15 may directly influence the expression of T-cell surface markers. Alternatively, T-ALL cells that resemble a mature thymocyte may be less dependent on high expression levels of MAP3K7 and thus less vulnerable to deletions of 6q15 in comparison to cortical or even less mature T-ALLs. No other clinical features were correlated with deletions of MAP3K7. Importantly, MAP3K7 deletions neither predicted treatment response nor the risk of relapse. Previous studies identified several mostly overlapping regions on chr 6q that are recurrently deleted in pediatric T-ALL and T-lymphoblastic lymphoma (Fig. 1, [7]). The centromeric region is localized on 6q14 and 6q15 and involves, among others, MAP3K7 and CASP8AP2. In a smaller cohort of patients, this deletion has been associated with a poor response to induction treatment [4]. However, we neither found an association of MAP3K7/CASP8AP2 deletions with an unfavorable treatment response nor with the cumulative incidence of relapse. Obviously, deletions of MAP3K7 are not a useful prognostic marker in the context of ALL-BFM 2000 and AIEOP-BFM ALL 2009 protocols. Future studies investigating the prognostic relevance of deletions on chr6q in T-ALL will ideally use high resolution mapping of copy number alterations in a large cohort of patients. Our work extends earlier findings that suggest that the effect of MAP3K7 deletions on the prognosis of malignancies is highly dependent on the cell of origin: MAP3K7 deletions are associated with advanced high-risk disease in prostate cancer [16, 27], but with a good prognosis in esophageal squamous cell carcinoma [49]. The effect on the prognosis of pediatric T-ALL seems to be very limited with current treatment protocols.
Transduction of T-ALL cell lines by AAV was efficient and resulted in transgene delivery efficiencies exceeding those typically reached by chemical transfection [38]. The simulation of MAP3K7 deletion by AAV-mediated shRNA depletion resulted in slower proliferation and increased apoptosis in all cell lines analyzed. The specificity of the effect of MAP3K7 depletion was confirmed with three different shRNA constructs in three different cell lines. Of note, the restraints in insert size in our AAV vectors did not allow rescue experiments reversing the effects of the depletion by ectopic expression of MAP3K7, so we cannot fully exclude off target effects.
It has previously been shown that MAP3K7 is indispensable for thymocyte and T-cell development [24, 50]. Similar observations in different cancer models have primarily been attributed to an inactivation of the NF-κB pathway upon MAP3K7 inhibition [25, 29]. For instance, chemical inhibition of MAP3K7 completely abolished NF-κB activation in AML cells, leading to cell death by apoptosis and decreased expression of IL8 [32]. The expression of a constitutively activated NF-κB p65 subunit only partially reduced these effects, indicating that MAP3K7 signaling was not restricted to NF-κB activation [32]. Similarly, our results neither showed an effect on IκB levels nor on NF-κB target gene expression as a response to MAP3K7 depletion. We conclude that the biologic effects of MAP3K7 depletion were independent of the NF-κB pathway. Several genes recurrently mutated in T-ALL influence the MAP3K7/NF-κB signaling pathway. Most prominently, NOTCH1 directly interacts with NF-κB proteins and the IKK complex, leading to their activation [51]. Both MOLT-4 and CCRF-CEM cell lines carry NOTCH1 mutations [52], and MAP3K7 might not be required additionally to activate NF-κB and the IKK complex. Furthermore, the function of MAP3K7 may not only depend on mutations in genes that can activate or inactivate NF-κB, but also on the cell type and the maturation stage. Specifically, MAP3K7 has a critical role for NF-κB activation in naïve T-cells, but is dispensable in effector T-cells [53]. This observation may also explain why deletions of MAP3K7 are found more frequently among leukemias with a more mature immunophenotype: Possibly “mature” lymphoblasts - in contrast to lymphoblasts resembling less mature precursors - do not depend on MAP3K7 for the activation of the NF-κB pathway, which has been shown to be required for leukemogenesis in several models [51, 54,55,56]. As we did not find a direct effect of MAP3K7 depletion on the activity of NF-κB, we suggest that MAP3K7 has an effect on cell proliferation that is independent from NF-κB.
Notably, our data do not formally exclude that the low MAP3K7 protein levels remaining after AAV-mediated depletion have different biological effects than the higher MAP3K7 protein levels remaining in cells carrying heterozygous deletions. We never observed homozygous deletions of MAP3K7 or the combination of a heterozygous deletion and a mutational inactivation of the remaining allele, indicating that some residual MAP3K7 activity is required for survival and proliferation of T-ALL cells. Although our results do not argue for a biological effect of monoallelic deletions of MAP3K7 in T-ALL, the fact that T-ALL cells require residual expression of MAP3K7 may imply the potential of MAP3K7 as a new treatment target: If MAP3K7 is not a tumor suppressor but co-deleted with another, yet to be identified tumor suppressor on 6q15, it may behave as a “CYCLOPS” gene [57]. Accordingly, MAP3K7 deletion may render leukemia cells highly vulnerable to inactivation of the remaining MAP3K7 allele. Possible candidate drugs that may be able to exploit this potential Achilles’ heel are chemical MAP3K7 inhibitors like (5Z)-7-Oxozeaenol, LYTAK1, AZ-TAK1 and NG52, which showed anti-tumor effects in various in vivo and in vitro cancer models but do not appear suitable for the use in a clinical setting due to low selectivity [58]. However, the multikinase inhibitor sorafenib [59], which has already been proven to be clinically effective in the treatment of various malignancies, inhibits MAP3K7 and may specifically target leukemias with low expression of MAP3K7 [60, 61].
Conclusions
Our results show that heterozygous MAP3K7 deletions are recurrently found in T-ALL patients, but do not affect patients’ outcome in the context of ALL-BFM treatment protocols. On a cellular level, MAP3K7 is essential for rapid proliferation and inhibition of apoptosis. In contrast to previous observations, we did not find inactivation of NF-κB to be the mechanism underlying the biological effects of MAP3K7 inactivation in T-ALL cell lines. The complete absence of homozygous MAP3K7 deletions and the proliferation arrest after efficient depletion indicate that MAP3K7 may be indispensable for T-ALL cells and thus a potential target for treatment.
Abbreviations
- AAV:
-
Adeno-associated virus
- GFP:
-
Green fluorescent protein
- MLPA:
-
Multiplex ligation-dependent probe amplification
- MOI:
-
Multiplicity of infection
- MRD:
-
Minimal residual disease
- PBS:
-
Phosphate-buffered saline
- pCIR:
-
Estimated cumulative incidence of relapse
- PCR:
-
Polymerase chain reaction
- PE:
-
R-Phycoerythrin
- pEFS:
-
Probability of 5-year event free survival
- PFA:
-
Paraformaldehyde
- qRT-PCR:
-
Quantitative Real-Time PCR
- RMD:
-
Region of minimal deletion
- shRNA:
-
Short hairpin RNA
- TAK1:
-
TGF-beta activated kinase 1
- T-ALL:
-
T-cell acute lymphoblastic leukemia
- TBS-T:
-
Tris-buffered saline with 0.5% Tween-20
- T-LBL:
-
T-cell lymphoblastic lymphoma
References
Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119(1):34–43.
Schrappe M, Reiter A, Ludwig WD, Harbott J, Zimmermann M, Hiddemann W, Niemeyer C, Henze G, Feldges A, Zintl F, et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. German-Austrian-Swiss ALL-BFM study group. Blood. 2000;95(11):3310–22.
Schrappe M, Valsecchi MG, Bartram CR, Schrauder A, Panzer-Grumayer R, Moricke A, Parasole R, Zimmermann M, Dworzak M, Buldini B, et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118(8):2077–84.
Remke M, Pfister S, Kox C, Toedt G, Becker N, Benner A, Werft W, Breit S, Liu S, Engel F, et al. High-resolution genomic profiling of childhood T-ALL reveals frequent copy-number alterations affecting the TGF-beta and PI3K-AKT pathways and deletions at 6q15-16.1 as a genomic marker for unfavorable early treatment response. Blood. 2009;114(5):1053–62.
Lopez-Nieva P, Vaquero C, Fernandez-Navarro P, Gonzalez-Sanchez L, Villa-Morales M, Santos J, Esteller M, Fernandez-Piqueras J. EPHA7, a new target gene for 6q deletion in T-cell lymphoblastic lymphomas. Carcinogenesis. 2012;33(2):452–8.
Sinclair PB, Sorour A, Martineau M, Harrison CJ, Mitchell WA, O'Neill E, Foroni L. A fluorescence in situ hybridization map of 6q deletions in acute lymphocytic leukemia: identification and analysis of a candidate tumor suppressor gene. Cancer Res. 2004;64(12):4089–98.
Burkhardt B, Moericke A, Klapper W, Greene F, Salzburg J, Damm-Welk C, Zimmermann M, Strauch K, Ludwig WD, Schrappe M, et al. Pediatric precursor T lymphoblastic leukemia and lymphoblastic lymphoma: differences in the common regions with loss of heterozygosity at chromosome 6q and their prognostic impact. Leuk Lymphoma. 2008;49(3):451–61.
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446(7137):758–64.
Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, Hoogerbrugge PM. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007;21(6):1258–66.
Paulsson K, Forestier E, Lilljebjorn H, Heldrup J, Behrendtz M, Young BD, Johansson B. Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2010;107(50):21719–24.
Ohshima K, Haraokaa S, Ishihara S, Ohgami A, Yoshioka S, Suzumiya J, Kikuchi M. Analysis of chromosome 6q deletion in EBV-associated NK cell leukaemia/lymphoma. Leuk Lymphoma. 2002;43(2):293–300.
Thelander EF, Ichimura K, Corcoran M, Barbany G, Nordgren A, Heyman M, Berglund M, Mungall A, Rosenquist R, Collins VP, et al. Characterization of 6q deletions in mature B cell lymphomas and childhood acute lymphoblastic leukemia. Leuk Lymphoma. 2008;49(3):477–87.
Kimm LR, deLeeuw RJ, Savage KJ, Rosenwald A, Campo E, Delabie J, Ott G, Muller-Hermelink HK, Jaffe ES, Rimsza LM, et al. Frequent occurrence of deletions in primary mediastinal B-cell lymphoma. Genes Chromosomes Cancer. 2007;46(12):1090–7.
Mohamed AN, Bentley G, Bonnett ML, Zonder J, Al-Katib A. Chromosome aberrations in a series of 120 multiple myeloma cases with abnormal karyotypes. Am J Hematol. 2007;82(12):1080–7.
Lee B, Yoon K, Lee S, Kang JM, Kim J, Shim SH, Kim HM, Song S, Naka K, Kim AK, et al. Homozygous deletions at 3p22, 5p14, 6q15, and 9p21 result in aberrant expression of tumor suppressor genes in gastric cancer. Genes Chromosomes Cancer. 2015;54(3):142–55.
Kluth M, Hesse J, Heinl A, Krohn A, Steurer S, Sirma H, Simon R, Mayer PS, Schumacher U, Grupp K, et al. Genomic deletion of MAP3K7 at 6q12-22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod Pathol. 2013;26(7):975–83.
Gorringe KL, Ramakrishna M, Williams LH, Sridhar A, Boyle SE, Bearfoot JL, Li J, Anglesio MS, Campbell IG. Are there any more ovarian tumor suppressor genes? A new perspective using ultra high-resolution copy number and loss of heterozygosity analysis. Genes Chromosomes Cancer. 2009;48(10):931–42.
Blaker H, Mechtersheimer G, Sutter C, Hertkorn C, Kern MA, Rieker RJ, Penzel R, Schirmacher P, Kloor M. Recurrent deletions at 6q in early age of onset non-HNPCC- and non-FAP-associated intestinal carcinomas. Evidence for a novel cancer susceptibility locus at 6q14-q22. Genes Chromosomes Cancer. 2008;47(2):159–64.
Kunz JB, Rausch T, Bandapalli OR, Eilers J, Pechanska P, Schuessele S, Assenov Y, Stutz AM, Kirschner-Schwabe R, Hof J, et al. Pediatric T-cell lymphoblastic leukemia evolves into relapse by clonal selection, acquisition of mutations and promoter hypomethylation. Haematologica. 2015;100(11):1442–50.
Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270(5244):2008–11.
Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6(11):1087–95.
Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress-induced rapid and transient activation of transforming growth factor-beta-activated kinase 1 (TAK1) in a signaling complex containing TAK1-binding protein TAB1 and TAB2. J Biol Chem. 2005;280(8):7359–68.
Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–10.
Liu HH, Xie M, Schneider MD, Chen ZJ. Essential role of TAK1 in thymocyte development and activation. Proc Natl Acad Sci U S A. 2006;103(31):11677–82.
Lamothe B, Lai Y, Hur L, Orozco NM, Wang J, Campos AD, Xie M, Schneider MD, Lockworth CR, Jakacky J, et al. Deletion of TAK1 in the myeloid lineage results in the spontaneous development of myelomonocytic leukemia in mice. PLoS One. 2012;7(12):e51228.
Ajibade AA, Wang HY, Wang RF. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013;34(7):307–16.
Liu W, Chang BL, Cramer S, Koty PP, Li T, Sun J, Turner AR, Von Kap-Herr C, Bobby P, Rao J, et al. Deletion of a small consensus region at 6q15, including the MAP3K7 gene, is significantly associated with high-grade prostate cancers. Clin Cancer Res. 2007;13(17):5028–33.
Wu M, Shi L, Cimic A, Romero L, Sui G, Lees CJ, Cline JM, Seals DF, Sirintrapun JS, McCoy TP, et al. Suppression of Tak1 promotes prostate tumorigenesis. Cancer Res. 2012;72(11):2833–43.
Safina A, Ren MQ, Vandette E, Bakin AV. TAK1 is required for TGF-beta 1-mediated regulation of matrix metalloproteinase-9 and metastasis. Oncogene. 2008;27(9):1198–207.
Safina A, Sotomayor P, Limoge M, Morrison C, Bakin AV. TAK1-TAB2 signaling contributes to bone destruction by breast carcinoma cells. Mol Cancer Res. 2011;9(8):1042–53.
Ray DM, Myers PH, Painter JT, Hoenerhoff MJ, Olden K, Roberts JD. Inhibition of transforming growth factor-beta-activated kinase-1 blocks cancer cell adhesion, invasion, and metastasis. Br J Cancer. 2012;107(1):129–36.
Bosman MC, Schepers H, Jaques J, Brouwers-Vos AZ, Quax WJ, Schuringa JJ, Vellenga E. The TAK1-NF-kappaB axis as therapeutic target for AML. Blood. 2014;124(20):3130–40.
Grimm D. Production methods for gene transfer vectors based on adeno-associated virus serotypes. Methods. 2002;28(2):146–57.
Borner K, Niopek D, Cotugno G, Kaldenbach M, Pankert T, Willemsen J, Zhang X, Schurmann N, Mockenhaupt S, Serva A, et al. Robust RNAi enhancement via human Argonaute-2 overexpression from plasmids, viral vectors and cell lines. Nucleic Acids Res. 2013;41(21):e199.
Kunze C, Borner K, Kienle E, Orschmann T, Rusha E, Schneider M, Radivojkov-Blagojevic M, Drukker M, Desbordes S, Grimm D, et al. Synthetic AAV/CRISPR vectors for blocking HIV-1 expression in persistently infected astrocytes. Glia. 2018;66(2):413–27.
Kalbfleisch JD, Prentice RL. The statistical analysis of failure time data. New York: Wiley; 1980.
Richter-Pechanska P, Kunz JB, Hof J, Zimmermann M, Rausch T, Bandapalli OR, Orlova E, Scapinello G, Sagi JC, Stanulla M, et al. Identification of a genetically defined ultra-high-risk group in relapsed pediatric T-lymphoblastic leukemia. Blood Cancer J. 2017;7(2):e523.
Palchetti S, Pozzi D, Marchini C, Amici A, Andreani C, Bartolacci C, Digiacomo L, Gambini V, Cardarelli F, Di Rienzo C, et al. Manipulation of lipoplex concentration at the cell surface boosts transfection efficiency in hard-to-transfect cells. Nanomedicine. 2017;13(2):681–91.
Inokuchi S, Aoyama T, Miura K, Osterreicher CH, Kodama Y, Miyai K, Akira S, Brenner DA, Seki E. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A. 2010;107(2):844–9.
Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20(50):7342–51.
Duyao MP, Buckler AJ, Sonenshein GE. Interaction of an NF-kappa B-like factor with a site upstream of the c-myc promoter. Proc Natl Acad Sci U S A. 1990;87(12):4727–31.
Xia D, Srinivas H, Ahn YH, Sethi G, Sheng X, Yung WK, Xia Q, Chiao PJ, Kim H, Brown PH, et al. Mitogen-activated protein kinase kinase-4 promotes cell survival by decreasing PTEN expression through an NF kappa B-dependent pathway. J Biol Chem. 2007;282(6):3507–19.
Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. 1990;171(1):35–47.
Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10(4):1498–506.
Chan H, Bartos DP, Owen-Schaub LB. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50-p65 recruitment. Mol Cell Biol. 1999;19(3):2098–108.
Singh NP, Nagarkatti M, Nagarkatti PS. Role of dioxin response element and nuclear factor-kappaB motifs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression. Mol Pharmacol. 2007;71(1):145–57.
Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1(1):75–87.
D'Angio M, Valsecchi MG, Testi AM, Conter V, Nunes V, Parasole R, Colombini A, Santoro N, Varotto S, Caniglia M, et al. Clinical features and outcome of SIL/TAL1-positive T-cell acute lymphoblastic leukemia in children and adolescents: a 10-year experience of the AIEOP group. Haematologica. 2015;100(1):e10–3.
Shi ZZ, Shang L, Jiang YY, Shi F, Xu X, Wang MR, Hao JJ. Identification of genomic biomarkers associated with the clinicopathological parameters and prognosis of esophageal squamous cell carcinoma. Cancer Biomark. 2015;15(6):755–61.
Sato S, Sanjo H, Tsujimura T, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Takeuchi O, Akira S. TAK1 is indispensable for development of T cells and prevention of colitis by the generation of regulatory T cells. Int Immunol. 2006;18(10):1405–11.
Vilimas T, Mascarenhas J, Palomero T, Mandal M, Buonamici S, Meng F, Thompson B, Spaulding C, Macaroun S, Alegre ML, et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. 2007;13(1):70–7.
Weng AP, Ferrando AA, Lee W, JPt M, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269–71.
Wan YY, Chi H, Xie M, Schneider MD, Flavell RA. The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat Immunol. 2006;7(8):851–8.
Carrasco D, Rizzo CA, Dorfman K, Bravo R. The v-rel oncogene promotes malignant T-cell leukemia/lymphoma in transgenic mice. EMBO J. 1996;15(14):3640–50.
Vacca A, Felli MP, Palermo R, Di Mario G, Calce A, Di Giovine M, Frati L, Gulino A, Screpanti I. Notch3 and pre-TCR interaction unveils distinct NF-kappaB pathways in T-cell development and leukemia. EMBO J. 2006;25(5):1000–8.
Portis T, Harding JC, Ratner L. The contribution of NF-kappa B activity to spontaneous proliferation and resistance to apoptosis in human T-cell leukemia virus type 1 tax-induced tumors. Blood. 2001;98(4):1200–8.
Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S, et al. Cancer vulnerabilities unveiled by genomic loss. Cell. 2012;150(4):842–54.
Santoro R, Carbone C, Piro G, Chiao PJ, Melisi D. TAK-ing aim at chemoresistance: the emerging role of MAP3K7 as a target for cancer therapy. Drug Resist Updat. 2017;33-35:36–42.
Zimmerman EI, Gibson AA, Hu S, Vasilyeva A, Orwick SJ, Du G, Mascara GP, Ong SS, Chen T, Vogel P, et al. Multikinase inhibitors induce cutaneous toxicity through OAT6-mediated uptake and MAP3K7-driven cell death. Cancer Res. 2016;76(1):117–26.
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–34.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
You D, Xin J, Volk A, Wei W, Schmidt R, Scurti G, Nand S, Breuer EK, Kuo PC, Breslin P, et al. FAK mediates a compensatory survival signal parallel to PI3K-AKT in PTEN-null T-ALL cells. Cell Rep. 2015;10(12):2055–68.
Acknowledgements
We thank Ellen Wiedtke for technical assistance with the AAV vector production. We thank Margit Happich for excellent technical assistance and Paulina Richter-Pechanska and Obul Reddy Bandapalli for helpful discussions.
Funding
The authors would like to thank the following institutions for grants: German Consortium for Translational Cancer Research (DKTK), “Tour der Hoffnung”, Manfred Lautenschläger Stiftung, European Commission (FP7, ERA-NET on Translational Cancer Research, TRANSCALL to MUM), Cluster of Excellence CellNetworks (DFG, EXC81), German Center for Infection Research (DZIF, TTU HIV 04.803). DMCDS, JE and JBK received fellowships from the Heidelberg Research Center for Molecular Medicine. DMCDS and JE were financed by the The Heidelberg Biosciences International Graduate School of Molecular and Cellular Biology (HBIGS).
Availability of data and materials
The dataset supporting the conclusions of this article is available upon request from the corresponding author.
Author information
Authors and Affiliations
Contributions
DMCDS performed the laboratory work, data analysis and wrote the manuscript. JE performed laboratory work, contributed to data analysis and revised the manuscript. ASV and EO performed laboratory work for the study. MZ performed statistical analysis of patient data, MSch and MSt contributed patient samples, KB and DG helped in AAV design and production. MUM and AEK supervised the research, contributed to experimental discussion and reviewed the manuscript. JBK designed and coordinated the research and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the institutional review boards of the Hannover Medical School and University of Schleswig-Holstein. Informed consent was obtained in accordance with the declaration of Helsinki.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Supplemental Methods: Details an patient data, AAV vector production, shRNA vector design. Table S1. Primers used for qRT-PCR. Figure S1. Transduction of T-ALL with anti-MAP3K7 shRNA leads to an efficient knockdown. Figure S2. Plasmid map of pscAAV-CMV-GFP-U6-sh construct with anti-MAP3K7 shRNA. Figure S3. Transduction of T-ALL with anti-MAP3K7 shRNA induces apoptosis. (ZIP 3146 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cordas dos Santos, D.M., Eilers, J., Sosa Vizcaino, A. et al. MAP3K7 is recurrently deleted in pediatric T-lymphoblastic leukemia and affects cell proliferation independently of NF-κB. BMC Cancer 18, 663 (2018). https://doi.org/10.1186/s12885-018-4525-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-018-4525-0