
Abstract
Background
Approximately 10–15% of ovarian carcinomas (OC) are attributed to inherited susceptibility, the majority of which are due to mutations in BRCA1 or BRCA2 (BRCA1/2). These patients display superior clinical outcome, including enhanced sensitivity to platinum-based chemotherapy. Here, we seek to investigate whether BRCA1/2 status influences the response rate to single-agent pegylated liposomal doxorubicin (PLD) in high grade serous (HGS) OC.
Methods
One hundred and forty-eight patients treated with single-agent PLD were identified retrospectively from the Edinburgh Ovarian Cancer Database. DNA was extracted from formalin-fixed paraffin-embedded (FFPE) archival tumour material and sequenced using the Ion Ampliseq BRCA1 and BRCA2 panel. A minimum variant allele frequency threshold was applied to correct for sequencing artefacts associated with formalin fixation.
Results
A superior response rate to PLD was observed in patients with HGS OC who harboured variants likely to affect BRCA1 or BRCA2 function compared to the BRCA1/2 wild-type population (36%, 9 of 25 patients versus 12.1%, 7 of 58 patients; p = 0.016). An enhanced response rate was also seen in patients harbouring only the BRCA1 SNP rs1799950, predicted to be detrimental to BRCA1 function (50%, 3 of 6 patients versus 12.1%, 7 of 58 patients; p = 0.044).
Conclusions
These data demonstrate that HGS OC patients with BRCA1/2 variants predicted damaging to protein function experience superior sensitivity to PLD, consistent with impaired DNA repair. Further characterisation of rs1799950 is now warranted in relation to chemosensitivity and susceptibility to developing ovarian carcinoma.
Similar content being viewed by others


Background
Ovarian cancer represents a substantial cause of mortality worldwide, with over 21,000 cases diagnosed, accounting for over 14,000 deaths, per year in the United States alone [1]. The majority of cases are ovarian carcinomas (OCs), approximately 10–15% of which arise in patients with inherited genetic susceptibility to disease [2, 3]. It is now recognised that the histologically-defined subgroups of OC represent distinct disease entities both molecularly and clinically, with high grade serous (HGS) OC accounting for the majority of cases (around 70%) [4].
Germline mutations in the DNA repair genes BRCA1 and BRCA2 (BRCA1/2) are responsible for the majority of hereditary OC, and around 15–20% harbour germline or somatic BRCA1/2 defects [5, 6]. Mutational inactivation of BRCA1/2 renders tumours deficient in homologous recombination DNA damage repair (HRR) [7, 8]. BRCA1/2-associated OC patients experience superior clinical outcome, despite their propensity for developing visceral metastases and a tendency to present with HGS histology [9,10,11,12,13]. These tumours display superior response rates to multiple lines of platinum-based chemotherapy, as well as superior sensitivity to PARP inhibitors, consistent with HRR-deficiency and dependence upon error-prone non-homologous end joining (NHEJ) to repair therapy-induced DNA damage [9, 14].
Pegylated liposomal doxorubicin (PLD) is a doxorubicin formulation, liposome-encapsulated and pegylated to increase drug half-life and reduce cardiotoxicity [15, 16]. PLD is often used in OC treatment in the advanced-stage, relapsed disease setting, with reported response rates of around 15% when used as a single agent [17, 18]. One mechanism of action of PLD is the induction of single-stranded and double-stranded DNA breaks through both free radical formation and direct intercalation into DNA, interfering with topoisomerase II-mediated repair [19].
A phase II trial comparing the PARP inhibitor olaparib at two doses versus PLD in a population of BRCA1/2-mutant patients with recurrent OC showed a greater than expected objective response rate to PLD [20]. Because BRCA1/2 status is known to influence the response rate of patients to platinum-based chemotherapy, and induction of DNA damage is a common mechanism of action between PLD and platinum, we postulated that BRCA1/2 status may also influence the response rates to PLD.
Three previous studies have attempted to address this hypothesis, but these investigations have suffered several methodological limitations [21,22,23]. All three studies include a number of untested “presumed BRCA1/2 negative” OC patients in their wild-type comparator cohorts [21, 23]. Two studies included a significant number of patients treated with PLD in combination with other agents, most commonly platinum, which account for around half of the PLD-treated population in each study [21, 22]. One study limited BRCA1/2 sequencing to regions of known founder mutations [22], and all three studies compared PLD response in a histologically heterogeneous population. Furthermore, these studies have limited sequencing to germline material, despite the substantial number of OC known to display somatic mutational inactivation of BRCA1/2 [24, 25]. Given the known differential chemosensitivity of histological subtypes of OC [4], the clear potential for previous analyses to be confounded by superior response rate to co-administered platinum in BRCA1/2-associated OC [9], the limited predictive power of family history in predicting germline BRCA status in the presumed negative populations [26], and the known phenotypic overlap between germline and somatic BRCA1/2-inactivated OC [24, 27], there is a clear need for comparison of response rate to PLD monotherapy to tumour BRCA1/2 status in a histologically uniform OC cohort.
Here we present next generation sequencing (NGS) of tumour DNA from a cohort of OC patients treated with PLD monotherapy from a single centre in order to better interrogate the interaction between BRCA1/2 status and response to PLD.
Methods
Cohort identification and pathology review
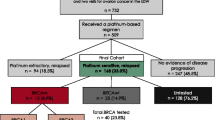
We retrospectively identified all patients treated with PLD monotherapy between 2001 and 2014 from the Edinburgh Ovarian Cancer Database (Fig. 1). 148 OC patients were identified. Of these, tumour material was available for translational research use in 119 cases. 10 μm sections were taken from archival tissue blocks alongside a 5 μm section to be stained with haemotoxylin and eosin (H&E). Ethical approval for the use of tumour material was obtained from South East Scotland Human Annotated Bioresource (East of Scotland Research Ethics Service Reference 10/S1402/33).

Flow diagram of HGS OC patients evaluable for PLD response
Tumour area identification and pathology review was conducted by an expert gynaecological pathologist using the H&E stained slide. Where histological subtype of OC was unclear from H&E alone, a combination of patient pathology reports and additional 5 μm sections immunohistochemically stained for WT1 and P53 proteins were used to determine OC histotype.
DNA extraction
H&E stained slides were used to guide macrodissection of four 10 μm FFPE tissue sections per specimen. DNA extraction was performed using the QIAamp DNA FFPE Tissue Kit and Deparaffinization Solution according to the manufacturer’s instructions.
NGS sequencing of BRCA1 and BRCA2 in FFPE-derived tumour DNA
Sequencing of the BRCA1 and BRCA2 coding regions was performed using the Ion Ampliseq BRCA1 and BRCA2 panel on the Ion Torrent sequencing platform. 111 patients were successfully sequenced for BRCA1 and BRCA2. BAM files were generated using Torrent Suite v4.6, and variants called using the Torrent Variant Caller v4.6.0.7. The minimum per-sample mean depth of coverage achieved was 916X; the median per-sample mean depth achieved was 4728X. The median uniformity of sequencing depth across targets was 90.5%. Called variants were functionally annotated using the Ensembl Variant Effect Predictor Version 75.
Sequencing of FFPE-derived DNA presents the challenges of both fragmentation and spontaneous deamination of DNA associated with formalin fixation [28, 29]. Consistent with these fixation artefacts, we observed a bias in the mutation spectrum of bi-allelic single nucleotide variants (SNVs) in our study compared to those reported in OC samples in the TCGA dataset, which utilised fresh frozen material (Fig. 2a) [25]. Consistent with previous reports, the strongest bias was in cytosine to thymine SNVs, likely as a result of cytosine deamination [29, 30].

a Comparison of bi-allelic SNV spectra between DNA extracted from FFPE and fresh frozen material in the TCGA data. b Proportions of previously documented variants retained (DVR) and novel variants removed (NVR) at various minimum allele frequency (AF) thresholds
To compensate for these artefacts, we applied minimum allele frequency (AF) thresholding to the set of variants. Comparing the proportion of previously documented (more likely true) variants retained to the proportion of novel (more likely false) variants, we found that for minimum AF > 10% more previously documented variants were lost than novel variants retained (Fig. 2b). We also compared the mutation spectrum of all retained bi-allelic SNVs at each AF threshold to the mutation spectrum from the fresh frozen TCGA samples, showing that the majority of the bias was removed at AF ≥ 10% (Table 1 and Additional file 1: Figure S1).
Together, these analyses demonstrated that a 10% AF threshold removed a large proportion of likely erroneous variants, while conserving the majority of likely true variants and minimising the difference in mutation spectrum compared to the TCGA data. Accordingly, variants detected at AF < 10% were discarded prior to analysis.
Classification of functionally relevant BRCA1 and BRCA2 variants
Frameshift and nonsense variants in BRCA1 and BRCA2 were classified as likely damaging to protein function, as were previously reported missense mutations with known pathogenicity. Splice site variants with reported pathogenicity were also classified as likely to be damaging. Missense mutations predicted as unlikely to affect protein function by both Sorting Intolerant From Tolerant (SIFT) and PolyPhen scores were discarded as non-functional variants, while those predicted likely deleterious by both were classified as likely to be damaging [31, 32]. Missense mutations with conflicting SIFT and PolyPhen predictions were discarded as variants of unknown significance.
Three insertion/deletion (indel) variants called at high frequency across the cohort were identified as suspected recurrent sequencing errors around homopolymer regions. Sanger sequencing of these regions in the respective tumours confirmed these as sequencing errors, consistent with previous reports of false-positive indel calling around problematic genomic regions on Ion Torrent NGS platforms (Additional file 2: Table S1) [33].
PLD response data
Patient response data were obtained retrospectively from the Edinburgh Ovarian Cancer Database. Responders were defined as patients who showed partial or complete CA125 tumour marker response or radiological response (either WHO or RECIST criteria as some patients predated RECIST reporting) from the PLD chemotherapy package. Patients who experienced stable disease, disease progression, or succumbed to disease on therapy were classified as non-responders. Patients for whom both CA125 data and scans were not available, or who received fewer than two cycles of PLD, were considered unevaluable for PLD response (Fig. 1).
Results
BRCA1 and BRCA2 mutation frequency
Among the 111 successfully sequenced PLD-treated patients, 46 variants likely to affect protein function were detected, comprising 26 BRCA1 variants and 20 BRCA2 variants. Of the BRCA1 variants, 12 were frameshift-inducing indels and 14 were missense variants, including 11 instances of the missense-causing SNP rs1799950 conferring a Gln356Arg amino-acid change and predicted to be detrimental to BRCA1 function by both SIFT and PolyPhen. Of the BRCA2 variants, 10 were frameshift indels, 7 were missense variants, 1 was a nonsense mutation and 2 were splice site variants.
Across the study cohort 31.5% (35 of 111) of patients harboured at least one variant in BRCA1 or BRCA2 predicted as likely to affect protein function (BRCA1/2-aberrant). Specifically, 20.7% (23 of 111 patients) harboured at least one variant in BRCA1 alone, 9.9% (11 of 111) displayed at least one variant in BRCA2 alone, and 0.9% (1 of 111 patients) harboured variants in both BRCA1 and BRCA2, consistent with previous reports of the higher BRCA1 mutation frequency in OC versus BRCA2 [10].
Of the BRCA1/2-aberrant population, 97.1% (34 of 35) were HGS OC, consistent with previous reports of the association between BRCA1/2 mutation and HGS histology. The remaining case was high grade endometrioid OC.
Patient demographics of BRCA1/2-aberrant and BRCA1/2 wild-type populations
There was no difference in FIGO stage at diagnosis, success of primary surgical tumour debulking, platinum sensitivity at PLD therapy initiation, or in the number of lines of cytotoxic chemotherapy received prior to PLD between the BRCA1/2-aberrant and wild-type groups (Table 2). BRCA1/2-aberrant patients were significantly younger at diagnosis compared with wild-type (median 55 years vs. 64 years, respectively; Welch Two-Sample t-test p < 0.001), consistent with previous associations of BRCA1/2 mutation with younger age at diagnosis [34].
PLD therapy response rate
78.4% (87 of 111) of patients were evaluable for response to PLD. 19.5% (17 of 87) were classified as responders by virtue of achieving either a CA125 or radiological response. This observed response rate is comparable to that reported in other studies investigating the use of single agent PLD in the advanced-stage recurrent disease setting [17, 18, 35].
Different histological subtypes of OC are known to display distinct response profiles to chemotherapy [36,37,38]. The vast majority of patients classified as responders had disease of HGS histology (94.1%, 16 of 17), giving a response rate of 19.3% (16 of 83) in the HGS population. Given the limited number of non-HGS patients evaluable for PLD response in this cohort (N = 4), comparison of differential response rates to PLD between histological OC groups could not be made.
BRCA1/2 status influences response rate to PLD in HGS OC
We observed a significantly higher response rate to PLD in the BRCA1/2-aberrant HGS OC population compared with the wild-type group in this study (Fig. 3) (36.0%, 9 of 25 patients vs. 12.1%, 7 of 58 patients; Fisher’s exact test p = 0.016). Of the 9 responses in the BRCA1/2-aberrant population, two were radiological, 6 were CA125 tumour marker response, and one was both radiological and CA125 response. Of the 7 responses in the wild-type population, two were radiological, three were CA125 tumour marker response, and two were both radiological and CA125 tumour marker response. This BRCA1/2-aberrant population comprised both BRCA1/2-mutant patients and patients who harboured the predicted detrimental rs1799950 BRCA1 SNP. When considering patients harbouring only rs1799950, we observed a significantly superior response rate to PLD versus the wild-type population (50%, 3 of 6 patients vs. 12.1%, 7 of 58; Fisher’s exact test p = 0.044), despite the small numbers of patients in this group (N = 6). There was a similar trend for superior response rate to PLD in the BRCA1/2-mutant population following the exclusion of patients whose tumour harboured only rs1799950 (31.6%, 6 of 19 patients vs. 12.1%, 7 of 58 patients; Fisher’s exact test p = 0.075).

Differential response rate to PLD chemotherapy according to BRCA1/2 status of sequenced tumour material. * indicates p < 0.05
Discussion
Using NGS technology, we were able to sequence FFPE-derived DNA for the BRCA1 and BRCA2 genes in 111 PLD-treated patients at high sequencing depth. We observed a strong bias in the SNV spectrum of FFPE-derived DNA versus that reported by the TCGA study which utilised fresh frozen patient material, consistent with formalin fixation-induced artefacts known to occur in FFPE-derived DNA [29, 30]. We used a minimum allele frequency cut-off threshold for called variants to correct the mutation spectrum for these fixation artefacts. While this approach risks filtering a minority of true variants, we have demonstrated that it removes the bulk of fixation-associated artefacts, whilst retaining the vast majority of likely true positive variants, achieving a practical equipoise for variant filtering.
We detected 29 and 22 BRCA1 and BRCA2 sequence variants likely to affect protein function, respectively. Among these, we identified rs1799950, a BRCA1 SNP predicted to be deleterious to protein function by PolyPhen and SIFT prediction tools. This SNP results in a non-conservative amino acid change in BRCA1 protein: the charged residue arginine is incorporated in place of the uncharged residue glutamine at amino acid 356. A previous study of high-risk prostate cancer families found that the minor allele of rs1799950 was associated with an increased risk of developing prostate cancer (OR 2.25, 95% confidence interval 1.21–4.20), but its relevance to treatment response is unstudied [39]. Furthermore, homozygosity of the minor allele state has previously been associated with breast cancer risk in a population from Saudi Arabia [40].
Of the 83 HGS OC patients evaluable for response to PLD chemotherapy, 19.3% responded to PLD, concurring with observations in previous studies [17, 18, 35]. Despite the low number of responders (N = 16), we were able to show a significantly superior response rate in those harbouring BRCA1/2 aberrations predicted damaging to protein function, when compared to BRCA1/2 wild-type samples, of approximately 2.5-fold. This is consistent with the hypothesis that impaired HRR function renders OC sensitive to non-platinum DNA damaging agents such as PLD, as well as platinum-based chemotherapy.
A minority of patients harboured the BRCA1 SNP rs1799950 with no other detected BRCA1/2 defects. While this group were severely limited by size, we observed a greater than four-fold response rate in this population compared to the wild type population. Notably, this SNP was reported to possess a minor allele frequency of 0.0596 in European populations by the 1000 Genomes Project, and would therefore be considered a common variant according to population genetics conventions. These data suggest that rs1799950 is biologically significant in terms of response to cytotoxic chemotherapy, and further characterisation of this variant is now warranted. Future studies should seek to evaluate whether rs1799950, and other common BRCA1 and BRCA2 variants, modulate sensitivity to platinum and other DNA damaging agents in vitro, and address whether such variants convey inherited susceptibility to malignancy, particularly to OC and breast carcinoma.
Previous studies have shown that BRCA1-deficient OC are more likely to display high levels of tumour infiltrating lymphocytes (TILs) and display an enrichment of immune response genes [41]. OC with high levels of T cell infiltration have superior clinical outcome, thought to be secondary to an improved anti-tumoural immune response [42, 43]. Recent work has suggested that PLD may enhance the immune response in BRCA1-deficient tumours [44], and this may contribute to the improved benefit from PLD seen in BRCA1/2-aberrant tumours.
The higher response rate in BRCA1/2-aberrant patients presents an additional argument for prospective BRCA1 and BRCA2 sequencing in all OC patients. Given the low response rate to PLD in the BRCA1/2 wild-type population (12.1% in our cohort), alternative therapies could be considered for the treatment of patients who have had germline or somatic BRCA1 and BRCA2 sequencing and have not displayed functionally relevant genetic changes in either of these genes. In light of the high response rate observed in BRCA1/2-aberrant patients, PLD should be considered as an active treatment option in patients with known BRCA1/2 mutations.
Moving forward, the question remains as to whether this observed superior response rate extends to patients with defects in other components of the HRR pathway. In particular, detrimental variants in HRR genes known to be mutationally inactivated in a minority of hereditary OC – such as BRIP1, PALB2 and CHEK2 – may also predict response rate to PLD. Furthermore, the impact on PLD response rate, if any, of epigenetic silencing of BRCA1 via promoter methylation remains unstudied.
Conclusion
HGS OC patients displaying BRCA1/2 sequence aberrations predicted detrimental to BRCA1 or BRCA2 protein function display an increased response rate to PLD. Patients harbouring the common BRCA1 variant rs1799950 may also display a superior response rate to PLD. These data support the notion that HGS OC patients with BRCA1/2 mutations are more sensitive to non-platinum DNA damaging agents compared to their BRCA1/2 wild-type counterparts. The role of rs1799950 in chemotherapy sensitivity and predisposition to OC and BC warrants further investigation.
Abbreviations
- AF:
-
allele frequency
- FFPE:
-
formalin-fixed paraffin-embedded
- H&E:
-
haemotoxylin and eosin
- HGS:
-
high grade serous
- HRR:
-
homologous recombination DNA repair
- Indel:
-
insertion/deletion
- NGS:
-
next generation sequencing
- NHEJ:
-
non-homologous end joining
- OC:
-
ovarian carcinoma
- PLD:
-
pegylated liposomal doxorubicin
- SIFT:
-
Sorting Intolerant From Tolerant
- SNV:
-
single nucleotide variant
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29.
Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005;104(12):2807–16.
Malander S, Rambech E, Kristoffersson U, Halvarsson B, Ridderheim M, Borg A, et al. The contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancer. Gynecol Oncol. 2006;101(2):238–43.
Hollis RL, Gourley C. Genetic and molecular changes in ovarian cancer. Cancer Biol Med. 2016;13(2):236–47.
Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108(44):18032–7.
Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian ovarian cancer study group. J Clin Oncol. 2012;30(21):2654–63.
Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4(4):511–8.
Tutt A, Bertwistle D, Valentine J, Gabriel A, Swift S, Ross G, et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001;20(17):4704–16.
Tan DS, Rothermundt C, Thomas K, Bancroft E, Eeles R, Shanley S, et al. BRCAness syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26(34):5530–6.
Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA. 2012;307(4):382–90.
Candido-dos-Reis FJ, Song H, Goode EL, Cunningham JM, Fridley BL, Larson MC, et al. Germline mutation in BRCA1 or BRCA2 and ten-year survival for women diagnosed with epithelial ovarian cancer. Clin Cancer Res. 2015;21(3):652–7.
Gourley C, Michie CO, Roxburgh P, Yap TA, Harden S, Paul J, et al. Increased incidence of visceral metastases in scottish patients with BRCA1/2-defective ovarian cancer: an extension of the ovarian BRCAness phenotype. J Clin Oncol Off J Am Soc Clin Oncol. 2010;28(15):2505–11.
Piek JM, Torrenga B, Hermsen B, Verheijen RH, Zweemer RP, Gille JJ, et al. Histopathological characteristics of BRCA1- and BRCA2-associated intraperitoneal cancer: a clinic-based study. Familial Cancer. 2003;2(2):73–8.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. The Lancet Oncology. 2014;15(8):852–61.
Safra T, Muggia F, Jeffers S, Tsao-Wei DD, Groshen S, Lyass O, et al. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11(8):1029–33.
Gabizon A, Martin F. Polyethylene glycol-coated (pegylated) liposomal doxorubicin. Rationale for use in solid tumours. Drugs. 1997;54(Suppl 4):15–21.
Campos SM, Penson RT, Mays AR, Berkowitz RS, Fuller AF, Goodman A, et al. The clinical utility of liposomal doxorubicin in recurrent ovarian cancer. Gynecol Oncol. 2001;81(2):206–12.
Gordon AN, Granai CO, Rose PG, Hainsworth J, Lopez A, Weissman C, et al. Phase II study of liposomal doxorubicin in platinum- and paclitaxel-refractory epithelial ovarian cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2000;18(17):3093–100.
Gewirtz DAA. Critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–41.
Kaye SB, Lubinski J, Matulonis U, Ang JE, Gourley C, Karlan BY, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2012;30(4):372–9.
Safra T, Borgato L, Nicoletto MO, Rolnitzky L, Pelles-Avraham S, Geva R, et al. BRCA mutation status and determinant of outcome in women with recurrent epithelial ovarian cancer treated with pegylated liposomal doxorubicin. Mol Cancer Ther. 2011;10(10):2000–7.
Safra T, Rogowski O, Muggia FM. The effect of germ-line BRCA mutations on response to chemotherapy and outcome of recurrent ovarian cancer. International journal of gynecological cancer: official journal of the international gynecological cancer. Society. 2014;24(3):488–95.
Adams SF, Marsh EB, Elmasri W, Halberstadt S, Vandecker S, Sammel MD, et al. A high response rate to liposomal doxorubicin is seen among women with BRCA mutations treated for recurrent epithelial ovarian cancer. Gynecol Oncol. 2011;123(3):486–91.
Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–75.
Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15.
Moller P, Hagen AI, Apold J, Maehle L, Clark N, Fiane B, et al. Genetic epidemiology of BRCA mutations--family history detects less than 50% of the mutation carriers. European journal of cancer (Oxford, England: 1990). 2007;43(11):1713–7.
Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD, 2nd, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol Off J Am Soc Clin Oncol 2010;28(22):3570–3576.
Quach N, Goodman MF, Shibata D. Vitro mutation artifacts after formalin fixation and error prone translesion synthesis during PCR. BMC Clin Pathol. 2004;4(1):1.
Hofreiter M, Jaenicke V, Serre D, von Haeseler A, Paabo SDNA. Sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001;29(23):4793–9.
Chen G, Mosier S, Gocke CD, Lin MT, Eshleman JR. Cytosine deamination is a major cause of baseline noise in next-generation sequencing. Mol Diagn Ther. 2014;18(5):587–93.
Ng PC, Henikoff SSIFT. Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, et al. Performance comparison of benchtop high-throughput sequencing platforms. Nat Biotechnol. 2012;30(5):434–9.
Boyd J, Sonoda Y, Federici MG, Bogomolniy F, Rhei E, Maresco DL, et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA. 2000;283(17):2260–5.
Wilailak S, Linasmita VA. Study of pegylated liposomal doxorubicin in platinum-refractory epithelial ovarian cancer. Oncology. 2004;67(3–4):183–6.
Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, et al. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer. 2000;88(11):2584–9.
Schmeler KM, Sun CC, Bodurka DC, Deavers MT, Malpica A, Coleman RL, et al. Neoadjuvant chemotherapy for low-grade serous carcinoma of the ovary or peritoneum. Gynecol Oncol. 2008;108(3):510–4.
Hess V, A'Hern R, Nasiri N, King DM, Blake PR, Barton DP, et al. Mucinous epithelial ovarian cancer: a separate entity requiring specific treatment. J Clin Oncol Off J Am Soc Clin Oncol. 2004;22(6):1040–4.
Douglas JA, Levin AM, Zuhlke KA, Ray AM, Johnson GR, Lange EM, et al. Common variation in the BRCA1 gene and prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2007;16(7):1510–6.
Merdad A, Gari MA, Hussein S, Al-Khayat S, Tashkandi H, Al-Maghrabi J, et al. Characterization of familial breast cancer in Saudi Arabia. BMC genomics. 2015;16 Suppl 1:S3.
Clarke B, Tinker AV, Lee CH, Subramanian S, van de Rijn M, Turbin D, et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol. 2009;22(3):393–402.
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–13.
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–43.
Mantia-Smaldone G, Ronner L, Blair A, Gamerman V, Morse C, Orsulic S, et al. The immunomodulatory effects of pegylated liposomal doxorubicin are amplified in BRCA1--deficient ovarian tumors and can be exploited to improve treatment response in a mouse model. Gynecol Oncol. 2014;133(3):584–90.
Acknowledgements
We extend our thanks to the patients who contributed to this study, and to the Edinburgh Ovarian Cancer Database from which data were collected for this research. We would also like to thank the Edinburgh Wellcome Trust Clinical Research Facility, Western General Hospital, Edinburgh, for their NGS services, and the Nicola Murray Foundation for their generous support of the Nicola Murray Centre for Ovarian Cancer Research.
Funding
RH is funded through a Medical Research Council PhD fellowship. MC received funding from the Nicola Murray Foundation. Funding bodies did not influence the study design, manuscript preparation, data collection, analysis or interpretation.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
The study was conceived by RH, MC and CG and the methodology was determined by RH, AM, MC, MA, AS, CS, SH and CG. TR, MM and FN contributed to data collection. All co-authors contributed to the data collection, analysis and/or interpretation. RH produced the first draft of the manuscript which was subsequently commented upon and redrafted by all co-authors. All co-authors agreed to the final draft. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval for the use of tumour material was obtained from South East Scotland Human Annotated Bioresource (East of Scotland Research Ethics Service Reference 10/S1402/33). This approval was granted without the requirement for individual patient consent because patient follow-up data were gathered as part of routine care, and most patients included in the study were deceased.
Consent for publication
Not applicable.
Competing interests
MM has sat on advisory boards for Roche and delivered lectures for Boehringer Ingelheim. CG has sat on advisory boards for AstraZeneca, Nucana and Clovis and has delivered lectures for Roche and AstraZeneca. CG has also received research funding from AstraZeneca, Novartis and Aprea.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figure S1.
Sum of squares differences (SSD) between our SNV spectrum and the fresh frozen TCGA SNV spectrum at various allele frequency threshold for variant filtering. (DOCX 53 kb)
Additional file 2: Table S1.
Recurrently called variants confirmed as sequencing errors by Sanger sequencing. (DOCX 11 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hollis, R.L., Meynert, A.M., Churchman, M. et al. Enhanced response rate to pegylated liposomal doxorubicin in high grade serous ovarian carcinomas harbouring BRCA1 and BRCA2 aberrations. BMC Cancer 18, 16 (2018). https://doi.org/10.1186/s12885-017-3981-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-017-3981-2