
Abstract
Background
Propionic acidemia (PA) is a severe monogenic disorder characterized by a deficiency of the mitochondrial protein propionyl-CoA carboxylase (PCC) enzyme, which is caused by mutations in the PCCA or PCCB gene. Preconception carrier screening could provide couples with meaningful information for their reproductive options; however, it is not widely performed in China.
Case presentation
This report describes a case of dizygotic twin siblings conceived by in vitro fertilization (IVF) and diagnosed with propionic acidemia (PA). Their parents had no history of PA. Tandem mass spectrometry and urine gas chromatography/mass spectrometry (GC/MS) of the twin siblings revealed markedly elevated propionyl carnitine (C3), C3/C2, and 3-hydroxypropionate in the plasma and urine. Whole-exome sequencing was performed for the twin siblings. A homozygous missense mutation, c.2002G > A (p.Gly668Arg) in PCCA, was identified in the twin siblings. Sanger sequencing confirmed the homozygous mutation in the twin siblings and identified their parents as heterozygous carriers of the c.2002G > A mutation in PCCA. Both neonates in this case died. This is an emotionally and financially devastating outcome that could have been avoided with genetic carrier screening before conception. If couples are screened before IVF and found to be silent carriers, then reproductive options (such as preimplantation genetic diagnosis or prenatal diagnosis) can be offered to achieve a healthy newborn.
Conclusion
This case is a reminder to infertile couples seeking IVF that it is beneficial to clarify whether they are silent carriers before undergoing IVF.
Similar content being viewed by others


Background
Propionic acidemia (PA) is a rare, life-threatening metabolic disorder caused by a deficiency of the mitochondrial propionyl-CoA carboxylase (PCC) enzyme. PA has an estimated incidence of 1 per 100,000 to 150,000 [1]. The PCC enzyme catalyzes carboxylation of propionyl-CoA to D-methylmalonyl-CoA; therefore, PA is characterized by toxic accumulation of propionyl-CoA and metabolites of branched chain amino acid catabolism, such as 3-hydroxypropionic acid and propionyl carnitine (C3) in plasma and urine [1]. Most patients present with ketoacidosis, difficulty feeding, lethargy, seizures, encephalopathy, and failure to thrive during the neonatal period. Patients who live beyond the neonatal period may experience intellectual deficits, chronic kidney disease, cardiomyopathies, and pancreatitis [2, 3].
PCC is a heterododecamer composed of six propionyl-CoA carboxylase alpha subunits and six propionyl-CoA carboxylase beta subunits. The alpha and beta subunits of the PCC enzyme are encoded by the PCCA and PCCB genes, which map to chromosome 13q32.3 and chromosome 3q22.3, respectively. Mutations in PCCA or PCCB result in defects of the PCC enzyme, leading to PA. To date, more than 200 variants of the PCCA or PCCB genes have been identified [4, 5]. PA is inherited in an autosomal recessive manner. Missense mutations account for approximately 50% of the variations and are the most frequent defects in PCCA and PCCB [6].
Assisted reproductive technologies (ART), including in vitro fertilization (IVF), are used to overcome infertility in many families. The goal of IVF is not only to achieve pregnancy but also to take home a healthy baby. Mendelian diseases are individually rare; however, collectively, they account for approximately 20% of infant mortality cases and approximately 10% of pediatric hospitalizations [7, 8]. Preconception carrier screening has gained more recognition; however, it is not widely performed for ART patients in China. We report a case of dizygotic twin siblings conceived by in vitro fertilization (IVF) who died of PA. The homozygous missense mutation c.2002G > A (p.Gly668Arg) in PCCA was identified in the twin siblings using whole-exome sequencing. The parents had no history of PA but were unknown carriers of the c.2002G > A mutation in PCCA.
Case presentation
Two newborn dizygotic twin siblings were born to unrelated nonconsanguineous parents with normal karyotypes. The pregnancy was achieved by IVF because of the mother’s fertility problems. The father had normal documented semen parameters. The mother initially presented to our institution at age 28 years with a 1-year history of secondary infertility. She had two previous pregnancies that resulted in one spontaneous abortion and one abortive ectopic pregnancy. Her menstrual cycles were irregular, with intervals of 28–45 days. She was diagnosed with polycystic ovary syndrome and hypothyroidism at our reproductive center. Her hysterosalpingogram showed no abnormalities, but she experienced a second ectopic pregnancy after ovulation induction treatment in our reproductive center. Unilateral salpingectomy was conducted through laparoscopy. The couple decided to try IVF. GnRH antagonist protocols were performed, 21 oocytes were retrieved, 16 oocytes were fertilized, and 12 embryos were formed; however, only four good-quality day 3 embryos were frozen. No pregnancies resulted from the first frozen embryo transfer (FET) cycle during which 2 day 3 embryos were thawed. The second FET cycle involved transferring 2 day 3 embryos, which resulted in the birth of dizygotic twin siblings. The mother was 31 years old and the father was 36 years old at the time of delivery.
The patients were dizygotic twin siblings born at 38 weeks of gestation. The birth weight of the girl was 2630 g and the birth weight of the boy was 2130 g. The girl was 1 week old when she presented with difficulty feeding, coma, seizures, acidosis, respiratory failure, neonatal septicemia, and hyperammonemia. A urinary organic acid analysis revealed elevation of 3-hydroxypropionic acid. Tandem mass spectrometry showed that she had elevated C3 and C3/C2 (Table 1), indicating she had PA. Her twin brother presented with difficulty feeding, lethargy, acidosis, neonatal septicemia, and hyperammonemia at the age of 1 week. Elevated 3-hydroxypropionic acid in urine and elevated C3 and C3/C2 in plasma suggested that he had PA (Table 1).
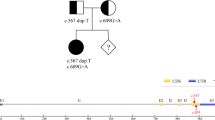
To identify the cause, whole-exome sequencing was performed for the twins by Guangzhou Kingmed Diagnostics Group Co., Ltd. Blood samples were collected from the patients, and genomic DNA was extracted using the QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany). The DNA libraries after enrichment and purification were sequenced on the NextSeq500 sequencer according to the manufacturer’s instructions (Illumina, San Diego, CA, USA). All reads were aligned to the reference human genome (UCSC hg19) using Burrows-Wheeler Aligner software (version 0.5.9-r16). The homozygous missense mutation c.2002G > A (p.Gly668Arg) in PCCA was identified in both twins; c.2002G > A is a known pathogenic mutation leading to PA. Furthermore, bioinformatic tools including PolyPhen 2, Sift, Mutation Taster, and MutPred score all classified this variation as pathogenic (Table S1). Sanger sequencing was performed for the two patients and their parents (Fig. 1). The parents had no history of PA; however, they were both identified as carriers of mutation c.2002G > A in PCCA. The girl died 24 h after hospitalization. Her twin brother died at age 1 month.

Sanger sequencing was performed to validate genotypes. Representative chromatograms of dizygotic twin siblings and their parents are shown. The arrows in the chromatograms indicate the location of the mutation (c.2002G > A)
Discussion and conclusions
We report dizygotic twin siblings conceived by IVF who both died of PA soon after birth. Genetic testing showed that the twins had a pathogenetic mutation in the PCCA gene and that their parents were unknown carriers of that PCCA mutation, thus confirming the diagnosis of PA. This type of event is mentally, emotionally, and financially devasting for families.
Variation c.2002G > A is located in exon 22 and causes an amino acid substitution at the 668 site of the PCCA protein. Gupta et al. screened PCCA and PCCB genes of 25 children in India with PA and identified that the c.2002G > A variation was associated with PA [9]. Bioinformatic tools classified this variation as pathogenic. This missense mutation c.2002G > A is in the biotinylation domain at the C-terminus. Campeau et al. examined this mutation (as Gly643Arg) in vitro to determine its capacity to support biotinylation and found that it decreased biotinylation by 95% of its initial value [10]. In this case report, the infertile couple was heterozygous for the missense mutation c.2002G > A; however, their genetic compositions were unknown before the delivery of their twins with PA.
Preconception carrier screening revealed that the average genomic carrier burden for severe pediatric recessive mutations was 2.8 [11]. Currently, because of technological advances like next-generation sequencing (NGS), preconception screening has become more feasible regardless of family history. According to recommendations (American College of Obstetrics and Gynecology Committee on Genetics), the disorders included in expanded carrier screening panels should: have a carrier frequency of 1 in 100 or greater, have a well-defined phenotype, have a detrimental effect on quality of life, cause cognitive or physical impairment, require surgical or medical intervention, or have an onset early in life [12]. The overall outcome of PA remains poor despite the existence of effective therapy such as a low-protein diet [1]. PA could be predicted through the usage of preconception genetic carrier screening and genetic counseling about reproductive options (such as preimplantation genetic diagnosis or prenatal diagnosis).
In the present study, twins with PA were conceived by an infertile couple through ART. Expanded preconception genetic carrier screening using NGS found that 5% of couples undergoing ART using their own gametes had pathogenic variants [13]. Researchers have estimated that preconception genetic carrier screening before ART for couples and patients requiring gamete donation results in prevention of 1.25% of newborns from being affected [13]. Franasiak et al. conducted expanded carrier screening for 3738 infertile couples and identified eight couples at risk of having an affected child, which indicated that expanded carrier screening affected the clinical decisions in 0.21% of cases associated with the infertile population [14]. Preconception carrier testing, ideally before pregnancy, is recommended by various professional societies [12, 15]. It is important to perform genetic screening of couples before ART to determine any genetic mutations so that steps (such as PGD) can be taken so that newborns conceived with ART are not affected. However, preconception carrier screening is not widely performed for ART patients in China. This case should be a reminder to infertile couples that it is critical to be aware of whether they are silent carriers before undergoing ART to reduce the risk of having an affected newborn. If two silent carriers are screened before IVF, a PGD procedure will be offered to achieve a genetically healthy newborn [16]. Further large-scale studies were needed to evaluate the benefits and the necessity of preconception carrier screening for infertile couples seeking ART.
Availability of data and materials
All datasets generated for this study are included in the article.
Abbreviations
- PA:
-
Propionic acidemia
- IVF:
-
In vitro fertilization
- PCC:
-
Propionyl-CoA carboxylase
- ART:
-
Assisted reproductive technologies
- FET:
-
Frozen embryo transfer
- PGD:
-
Preimplantation genetic diagnosis
- NGS:
-
Next-generation sequencing
References
Baumgartner MR, Horster F, Dionisi-Vici C, Haliloglu G, Karall D, Chapman KA, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9:130.
Haijes HA, Jans JJM, Tas SY, Verhoeven-Duif NM, van Hasselt PM. Pathophysiology of propionic and methylmalonic acidemias. Part 1: complications. J Inherit Metab Dis. 2019;42(5):730–44.
Shchelochkov OA, Manoli I, Sloan JL, Ferry S, Pass A, Van Ryzin C, et al. Chronic kidney disease in propionic acidemia. Genet Med. 2019;21(12):2830–5.
Wongkittichote P, Ah Mew N, Chapman KA. Propionyl-CoA carboxylase - a review. Mol Genet Metab. 2017;122(4):145–52.
Rivera-Barahona A, Navarrete R, Garcia-Rodriguez R, Richard E, Ugarte M, Perez-Cerda C, et al. Identification of 34 novel mutations in propionic acidemia: functional characterization of missense variants and phenotype associations. Mol Genet Metab. 2018;125(3):266–75.
Desviat LR, Sanchez-Alcudia R, Perez B, Perez-Cerda C, Navarrete R, Vijzelaar R, et al. High frequency of large genomic deletions in the PCCA gene causing propionic acidemia. Mol Genet Metab. 2009;96(4):171–6.
Costa T, Scriver CR, Childs B. The effect of Mendelian disease on human health: a measurement. Am J Med Genet. 1985;21(2):231–42.
Kumar P, Radhakrishnan J, Chowdhary MA, Giampietro PF. Prevalence and patterns of presentation of genetic disorders in a pediatric emergency department. Mayo Clin Proc. 2001;76(8):777–83.
Gupta D, Bijarnia-Mahay S, Kohli S, Saxena R, Puri RD, Shigematsu Y, et al. Seventeen novel mutations in PCCA and PCCB genes in Indian propionic Acidemia patients, and their outcomes. Genet Test Mol Biomarkers. 2016;20(7):373–82.
Campeau E, Dupuis L, Leon-Del-Rio A, Gravel R. Coding sequence mutations in the alpha subunit of propionyl-CoA carboxylase in patients with propionic acidemia. Mol Genet Metab. 1999;67(1):11–22.
Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, et al. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med. 2011;3(65):65ra64.
Committee on G: Committee Opinion No. 690: carrier screening in the age of genomic medicine. Obstet Gynecol. 2017;129(3):e35–40.
Martin J, Asan YY, Alberola T, Rodriguez-Iglesias B, Jimenez-Almazan J, Li Q, et al. Comprehensive carrier genetic test using next-generation deoxyribonucleic acid sequencing in infertile couples wishing to conceive through assisted reproductive technology. Fertil Steril. 2015;104(5):1286–93.
Franasiak JM, Olcha M, Bergh PA, Hong KH, Werner MD, Forman EJ, et al. Expanded carrier screening in an infertile population: how often is clinical decision making affected? Genet Med. 2016;18(11):1097–101.
Edwards JG, Feldman G, Goldberg J, Gregg AR, Norton ME, Rose NC, et al. Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of genetic counselors, perinatal Quality Foundation, and Society for Maternal-Fetal Medicine. Obstet Gynecol. 2015;125(3):653–62.
Alberola TM, Bautista-Llacer R, Vendrell X, Garcia-Mengual E, Pardo M, Vila M, et al. Case report: birth of healthy twins after preimplantation genetic diagnosis of propionic acidemia. J Assist Reprod Genet. 2011;28(3):211–6.
Acknowledgments
The authors thank Editage for English language editing.
Funding
This research was supported by research grants from the National Natural Science Foundation of China (81701410) and Beijing-Tianjin-HeBei project (19JCZDJC65000), Tianjin Science and Technology Project (20JCQNJC00470).
Author information
Authors and Affiliations
Contributions
X.B. supported the study; Y.T., G.W., and W.S collected all the clinical data and blood samples; Y.T interpreted the data and drafted the manuscript; X.B. and W.S revised the article. All authors gave their final approval for the version to be published.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Institutional Review Board of Tianjin Medical University General Hospital and written informed consent was obtained from the couple.
Consent for publication
Written consent for publication was obtained from the couple in this study.
Competing interests
All authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. PCCA variants identified in twin siblings with propionic academia.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tian, Y., Wang, G., Shi, W. et al. Propionic acidemia identified in twin siblings conceived by in vitro fertilization (IVF) with parents who were unknown carriers of a PCCA mutation. BMC Pregnancy Childbirth 20, 689 (2020). https://doi.org/10.1186/s12884-020-03391-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12884-020-03391-z