
Abstract
Background
Variants in the SLC25A1 gene are associated with a severe neurometabolic disease, D-2- and L-2-hydroxyglutaric aciduria (D/L-2-HGA). A report in 2014 presented the first account of congenital myasthenic syndrome (CMS) with mild intellectual disability (ID) caused by SLC25A1. To date, only two missense variants in SLC25A1 have been linked to CMS.
Case presentations
A Chinese boy presented fatigable muscular weakness, myasthenic crisis, epilepsy and developmental delay along with mild elevation of urinary 2-ketoglutarate (2-KG) and lactic acid levels. He showed a partial response to pyridostigmine. Genetic analysis using trio whole-exome sequencing (WES), Sanger sequencing, and cosegregation analyses revealed two novel pathogenic variants of SLC25A1 (c.628C > T, p.R210X; c.145G > A, p.V49M).
Conclusions
We report a boy who carries novel compound heterozygous variants of SLC25A1 and presents a phenotype intermediate between CMS and D/L-2-HGA. This case expands the range of known phenotypes and genotypes associated with SLC25A1.
Similar content being viewed by others


Background
Congenital myasthenic syndrome (CMS) is a heterogeneous group of disorders caused by mutations that lead to impairment of neuromuscular transmission, mainly by affecting the components of the neuromuscular junction (NMJ). These disorders are usually manifested as early-onset fatigable ocular, bulbar and limb muscle weakness. However, increasingly recognized, the recently discovered CMS genes result in some complex manifestations that make the diagnosis more difficult.
SLC25A1 encodes the mitochondrial citrate carrier SLC25A1 (CIC) [1]. Variants in the SLC25A1 gene are associated with a severe neurometabolic disease, D-2- and L-2-hydroxyglutaric aciduria (D/L-2-HGA), which is characterized clinically by severe developmental delay, hypotonia, and seizures [2,3,4]. In 2014, two British siblings presenting with mild intellectual disability (ID) had been reported firstly to be complicated by CMS due to a homozygous (c.740G > A; p. R247Q) missense variant in SLC25A1 [5]. Another four patients with the same missense variant in SLC25A1 were identified as having mild CMS with ID in 2019 [6]. In addition to the recurrent reported pathogenic variant (c.740G > A; p.R247Q), the other missense variant (c.205G > T; p. D69Y) was recently confirmed in three siblings with CMS [7].
To date, only two missense variants in SLC25A1 have been associated with CMS. We report a patient with an intermediate phenotype between CMS and D/L-2-HGA due to two novel compound heterozygous variants. We review the clinical and genetic features of CMS and further delineate the phenotype and genotype of this syndrome.
Case presentation
The patient was diagnosed and fully investigated through clinical information, an electrophysiological examination and muscle biopsy at the Children’s Hospital of Fudan University, Shanghai, China. The patient is an 8-year-old male, the second child of healthy nonconsanguineous Chinese parents who also have a healthy 15-year-old daughter. The boy was born at term in an uneventful delivery, with a birth weight of 3.9 kg. At birth, he was noticed to have laryngeal stridor and a quiet cry.
At midnight one night when the boy was 9 months old, he was found with blue lips and an unresponsive stare. After approximately 10 min, he became conscious and felt tired. This event was not initially considered to be epileptic, as the child’s electroencephalograms (EEGs) were normal. However, two similar episodes manifesting as focal impaired awareness seizures were noted at 1 year old and 6 years old. There were some focal epileptic discharges on the boy’s EEG after the second seizure. Because his seizures were not very frequent, we did not prescribe any antiepileptic drugs to him.
On follow-up, developmental delay was noted. The patient could control his head at 4.5 months, sit without support at 8 months, and walk independently at 2 years. He could speak at 16 months, but his speech was inarticulate. The Gesell Developmental Schedules were applied at 3 years and 2 months old, and his scores were 66, 54, 51, and 72 in the social, adaptive, language, and motor areas, respectively. At present, he has learning difficulties and is unable to receive mainstream schooling.
After 18 months of age, he was noticed to move his head rather than his eyes to look upward. He developed intermittent bilateral ptosis and occasional strabismus at 23 months; both conditions were aggravated by exercise and improved with rest. After 2 years, his parents found that he had an unsteady gait and fatigable weakness of the limbs and face.
The patient had two myasthenic crises during follow-up. At 4 years and 8 months, he was admitted to the emergency department for sudden dyspnoea and exacerbation of limb weakness following acute bronchitis. He was placed on mechanical ventilation and an antibiotic regimen for several days. In the winter of 2018, he was diagnosed with pneumonia that presented as a fever and cough. He did not improve after taking oral antibiotics for 6 days. He abruptly presented dyspnoea and dysphonia on admission. Eventually, he recovered through ventilator-assisted breathing. His lactate levels were only mildly elevated in both crises.
Examination revealed some facial weakness with mild to moderate bilateral ptosis and ophthalmoplegia. The patient also had fatigable limb weakness. He was hypotonic, and his reflexes were normal.
Basic laboratory parameters were normal, including blood count, hepatic and renal function, creatine kinase, amino acid profile and acylcarnitine profile assays. Two urinary organic acid analyses at 2 and 8 years old all revealed a mild elevation of 2-ketoglutarate (2-KG) and lactic acid. Four measurements of blood lactate from 2 to 8 years were slightly elevated, ranging from 2.5 to 4.0 mmol/L (reference range 0.7–2.1). The patient was negative for acetylcholine receptor (AChR) antibodies.
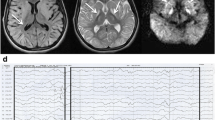
Echocardiograms, electrocardiograms, and brain MRI were normal. Muscle biopsy suggested atrophy of type IIB muscle fibres with normal cytochrome c oxidase (COX) and succinate dehydrogenase (SDH) staining, and the electron microscopy results were normal, with no mitochondrial changes (Supplementary data). Repetitive nerve stimulation of the axillary nerve and median nerve at 3 Hz showed 25.1 and 14.3% decreases in compound muscle action potentials (CMAPs), respectively.
The patient was given pyridostigmine at 3 years of age, although he had some other features unrelated to CMS. His parents reported that pyridostigmine was partially effective, but he did not improve with a further trial of salbutamol. The daily dosage of pyridostigmine was increased from the initial 45 mg to the current 180 mg. Initially, there was no improvement with 45 mg per day of pyridostigmine, and the dosage was gradually increased to 90 mg per day after 8 months. The parents said the boy’s ptosis was alleviated and his overall physical activity was increased. After 2 weeks of observation, salbutamol was added at a dosage of 2 mg per day. In August 2015, pyridostigmine and salbutamol were tapered off to determine the effect. During the three-week observation period, the patient showed more severe ptosis, dysphagia and proximal weakness. Both medications were reintroduced, and the dosage was gradually increased based on his weight. Salbutamol (4 mg per day) was stopped again until June 2017 to observe the effect. However, no differences were reported. Since then, the patient has taken pyridostigmine alone. He has not reported any adverse effects from pyridostigmine. Electrocardiography was routinely performed every 3 months when he was taking salbutamol; the results were all normal.
As the patient had some clinical manifestations of CMS, a neuromuscular genetic panel was first performed. The panel revealed two compound heterozygous missense variants in PLEC, which is related to epidermolysis bullosa simplex, although the boy had no history related to that condition. Subsequent genetic testing of the mitochondria from a blood sample was normal, with no evidence of point mutations or deletions/duplications. Then, whole-exome sequencing was performed, leading to the identification of the gene SLC25A1. The patient was compound heterozygous for the novel SLC25A1 variants c.628C > T (p.R210X) and c.145G > A (p.V49M). The father was heterozygous for c.628C > T, while the mother was heterozygous for c.145G > A. These variants were confirmed by Sanger sequencing (Fig. 1). Subsequent Sanger sequencing for 2 SLC25A1 variants in the sister showed that she was heterozygous for c.628C > T (p.R210X). The patient’s two SLC25A1 variants have never been reported in the 1000 Genomes Project or the ExAC database. According to the ACMG 2015 guidelines, the c.628C > T (p.R210X) variant, resulting in truncated proteins, was classified as pathogenic (PVS1, PM2, PP4), and the c.145G > A (p.V49M) variant was classified as likely pathogenic (PM2, PM3, PP3, PP4) [8].

Results of the Sanger sequencing. Electropherograms of the Sanger sequencing show the two heterozygous variants (c.145G > A on the left and c.628C > T on the right) in the patient. The mother was heterozygous for c.145G > A, while the father was heterozygous for c.628C > T. The sister was heterozygous for c.628C > T (p.R210X). The variants are indicated by the black arrows
Given the combination of his clinical phenotype (fatigable muscular weakness, myasthenic crisis, epilepsy, developmental delay, mild elevation of 2-KG and lactic acid in urine) and the molecular genetic findings, the proband was diagnosed with SLC25A1-related disorder. Moreover, we consider the patient’s phenotype to be intermediate between the reported phenotypes of CMS and D/L-2-HGA.
Sixteen patients with CMS due to SLC25A1 have been reported, including our patient (Table 1). All patients had very early onset. The myasthenic features consisted of ocular and proximal limb weakness in all patients, bulbar weakness in 6 patients, and cervical and facial involvement in two patients. Repetitive nerve stimulation (RNS) at 3 Hz elicited a positive response in four patients, and abnormal jitter on nerve block was observed in one patient. Partial responses to acetylcholinesterase (AChE) inhibitors were recorded in three patients, and a partial response to 3,4-diaminopyridine (3,4-DAP) was recorded in one patient. Of three patients, none was responsive to salbutamol. Mild ID was observed in 9 patients. To our knowledge, only 4 out of 27 variants in SLC25A1 are associated with CMS (Fig. 2).

Distribution of 27 novel and previously published SLC25A1 variants (including our case in red). The upper panel of the schematic shows all pathogenic variants in patients in whom CMS was the main feature, while the mutations in the lower panel correspond to the individuals who had been diagnosed with D/L-2-HGA
Discussion and conclusions
Here, we report that a typical CMS patient with some features of D/L-2-HGA harboured novel pathogenic variants in SLC25A1.
SLC25A1, located on chromosome 22q11, encodes the mitochondrial citrate carrier SLC25A1 (CIC). This protein belongs to the SLC25 family of mitochondrial carriers (MCs), which are mainly localized in the inner mitochondrial membrane and are responsible for the trafficking of a variety of metabolites [9, 10]. The locus of this gene (22q11.2) has also attracted some attention, as it is either translocated or amplified in some tumours and deleted in DiGeorge syndrome and possibly schizophrenia [11, 12]. Since 2013, SLC25A1 has been reported to be linked to human diseases. Pathogenic variants in SLC25A1 cause the combined D/L-2-HGA [2,3,4] and CMS phenotype (MIM) [5, 6] with a recessive mode of inheritance.
The phenotype of SLC25A1-related diseases should be a consecutive spectrum consisting of CMS, D/L-2-HGA and the intermediate type (with combined features of CMS and D/L-2-HGA). The diagnosis of congenital myasthenia is based on the presence of early-onset fatigable weakness, impaired NMJ transmission, absence of autoantibodies and response to treatment [13, 14]. Our patient has generalized fluctuating weakness, a positive response to RNS, and negative AChR antibodies, and he manifests some response to pyridostigmine. The myasthenic crises in our patient suggest that clinicians should take respiratory distress or bulbar exacerbation as a warning sign. In addition to the typical manifestation of CMS, the patient had some other features related to the other polarity of SLC25A1 deficiency, D/L-2-HGA. The boy had occasional epilepsy, hypotonia, and abnormal metabolic signs in the urine and blood.
The origin of the elevated D and L enantiomers of 2-HG due to SLC25A1 is still unclear, but one proposed explanation is the accumulation of citrate and other tricarboxylic acid (TCA) cycle intermediates in the mitochondria, including 2-KG, which is converted to D- and L-2-HG [15]. There was a slight elevation of 2-KG and lactate levels in our patient’s urine. In addition, two out of four patients in a previous study had increased lactate levels after exercise [6]. This result suggested that CMS due to SLC25A1 might involve a defect that mildly reduces mitochondrial function. Further studies are required to explain whether there is any correlation between NMJ defects and abnormal metabolite levels.
To our knowledge, only four mutations in SLC25A1 have been detected in CMS patients to date. Meanwhile, more than 20 pathogenic variants are reportedly related to D/L-2-HGA. What is the relationship between phenotypes and SLC25A1 genotypes? SLC25A1 knockdown zebrafish showed motility defects and NMJ abnormalities in less severely affected morphants, while additional defects of the brain and heart were observed in more severely affected morphants [5]. In 2018, phenotype–genotype correlation studies of SLC25A1, using the residual activity of missense variants compared to wild-type transfectants, showed that some patients with D/L-2-HGA harboured variants that caused a mild reduction in protein activity [16]. Thus, protein activity does not provide the full story.
In conclusion, we expand the range of known phenotypes and genotypes associated with SLC25A1-related disorders. The phenotypes of SLC25A1-related diseases seem to be a consecutive spectrum consisting of CMS, D/L-2-HGA and the intermediate type (combining features of CMS and D/L-2-HGA). Future functional studies of SLC25A1 are needed to delineate the phenotypic differences.
Availability of data and materials
The data used to support the findings of this study are available from the corresponding author upon request, without breaching participant confidentiality.
Abbreviations
- D/L-2-HGA:
-
D-2 and L-2-hydroxyglutaric aciduria
- CMS:
-
Congenital myasthenic syndrome
- ID:
-
Intellectual disability
- 2-KG:
-
2-ketoglutarate
- WES:
-
Whole-exome sequencing
- NMJ:
-
Neuromuscular junction
- ACMG:
-
American College of Medical Genetics
- EEG:
-
Electroencephalogram
- AChR:
-
Acetylcholine receptor
- COX:
-
Cytochrome c oxidase
- SDH:
-
Succinate dehydrogenase
- CMAP:
-
Compound muscle action potentials
- RNS:
-
Repetitive nerve stimulation
- AChE:
-
Acetylcholinesterase
- 3,4-DAP:
-
3,4-diaminopyridine
- TCA:
-
Tricarboxylic acid
- CIC:
-
Citrate carrier SLC25A1
- MC:
-
Mitochondrial carrier
References
Kaplan RS, Mayor JA, Wood DO. The mitochondrial tricarboxylate transport protein. cDNA cloning, primary structure, and comparison with other mitochondrial transport proteins. J Biol Chem. 1993;268:13682–90.
Nota B, Struys EA, Pop A, et al. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2- hydroxyglutaric aciduria. Am J Hum Genet. 2013;92:627–31.
Cohen I, Staretz-Chacham O, Wormser O, et al. A novel homozygous SLC25A1 mutation with impaired mitochondrial complex V: possible phenotypic expansion. Am J Med Genet A. 2018;176:330–6.
Smith A, McBride S, Marcadier JL, et al. Severe neonatal presentation of mitochondrial citrate carrier (SLC25A1) deficiency. JIMD Rep. 2016;30:73–9.
Chaouch A, Porcelli V, Cox D, et al. Mutations in the mitochondrial citrate carrier SLC25A1 are associated with impaired neuromuscular transmission. J Neuromuscul Dis. 2014;1:75–90.
Balaraju S, Töpf A, McMacken G, et al. Congenital myasthenic syndrome with mild intellectual disability caused by a recurrent SLC25A1 variant. Eur J Hum Genet. 2020;28:373–7.
Al-Futaisi A, Ahmad F, Al-Kasbi G, et al. Missense mutations in SLC25A1 are associated with congenital myasthenic syndrome type 23. Clin Genet. 2020;97:666–7.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Asp Med. 2013;34:465–84.
Palmieri F, Monné M. Discoveries, metabolic roles and diseases of mitochondrial carriers: a review. Biochim Biophys Acta. 2016;1863:2362–78.
Maynard TM, Meechan DW, Dudevoir ML, et al. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci. 2008;39:439–51.
Williams NM, Spurlock G, Norton N, et al. Mutation screening and LD mapping in the VCFS deleted region of chromosome 22q11 in schizophrenia using a novel DNA pooling approach. Mol Psychiatry. 2002;7:1092–100.
Hantai D, Nicole S, Eymard B. Congenital myasthenic syndromes: an update. Curr Opin Neurol. 2013;26:561–8.
Beeson D, Hantai D, Lochmuller H, et al. 126th international workshop: congenital myasthenic syndromes, 24-26 September 2004, Naarden, the Netherlands. Neuromuscul Disord. 2005;15:498–512.
Kranendijk M, Struys EA, Salomons GS, et al. Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35:571–87.
Pop A, Williams M, Struys EA, et al. An overview of combined D-2- and L-2-hydroxyglutaric aciduria: functional analysis of CIC variants. J Inherit Metab Dis. 2018;41:169–80.
Acknowledgements
We thank the patient and his family for participating in this case study.
Funding
No funding was received for this study.
Author information
Authors and Affiliations
Contributions
WH L wrote the case report and prepared the figures and table. SZ Z designed the study and substantively revised the manuscript. M Z, LM Z, YY S, L Z and XH L investigated and followed the patient. BB W performed the whole-exome sequencing. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Children’s Hospital of Fudan University, and written consent was obtained from the patient’s parents to publish his clinical and genetic data. Our study is in accordance with the Declaration of Helsinki and the CARE guidelines/methodology. All authors agree to the Publishing Statements of BMC Neurology.
Consent for publication
Written consent for publication was obtained from each of the family members. Written consent was obtained from the patient’s parents to publish his clinical and genetic data.
Competing interests
None of the authors has any competing interests or conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Muscle biopsy. A–D Skeletal muscle sections from the patient were stained with COX (A), HE(B), MGT2 (C) and SDH (D) to visualize mitochondria. E-F Electron microscopy findings.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, W., Zhang, M., Zhang, L. et al. A case report of an intermediate phenotype between congenital myasthenic syndrome and D-2- and L-2-hydroxyglutaric aciduria due to novel SLC25A1 variants. BMC Neurol 20, 278 (2020). https://doi.org/10.1186/s12883-020-01854-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-020-01854-6