
Abstract
Background
Neuronal intranuclear inclusion disease (NIID) is a rare neurodegenerative condition characterized by the loss of neurons and the presence of eosinophilic nuclear inclusions in the central and peripheral nervous system, skin and visceral organs. In this paper, we present a case of NIID with recurrent encephalitic attacks that remained stable and nonprogressive for seven years; no such case has previously been reported.
Case presentation
A 63-year-old female was hospitalized due to light-headedness, vomiting, unstable gait and cognitive impairment. Seven years prior, she had experienced an episode of light-headedness, central facial paralysis, unstable gait, aphasia, nausea, vomiting and loss of consciousness. She regained consciousness within 12 h, and her other symptoms were completely resolved within one week. During the present hospitalization, a brain magnetic resonance imaging (MRI) examination detected high signal intensity on diffusion-weighted imaging (DWI) of the bilateral frontal grey matter–white matter junction. We reviewed the patient’s previous MRI results and found that she had also had high signal intensity on DWI of the bilateral frontal grey matter–white matter junction seven years prior. In the intervening seven years, the high signal intensity in the frontal lobes had spread along the grey matter–white matter junction, but the deep white matter remained unaffected. Skin biopsy was performed, and intranuclear inclusions were found in adipocytes, fibroblasts and sweat gland cells. GGC repeat expansions in the NOTCH2NLC (Notch 2 N-terminal like C) gene confirmed the diagnosis of NIID. She received supportive treatment such as nutrition support therapy and vitamin B and C supplementation, as well as symptomatic treatment during hospitalization. The patient’s symptoms were completely relieved within one week.
Conclusion
This is a detailed report of a case of NIID with multiple reversible encephalitic attacks, diagnosed by clinical symptoms, intranuclear inclusions, characteristic DWI signals, and genetic tests.
Similar content being viewed by others


Background
NIID is a rare, slowly progressing neurodegenerative disease characterized by eosinophilic intranuclear hyaline inclusions in cells of the central and peripheral nervous system (including the autonomic nervous system) and the visceral organs [1]. GGC repeat expansion in the 5′ untranslated region (5′ UTR) of the NOTCH2NLC (Notch 2 N-terminal like C) gene is associated with the mechanism of NIID [2]. NIID may occur from infancy to old age. According to the age of onset, NIID can be categorized as infantile, juvenile, or adult. Sporadic NIID emerges between 51 and 76 years of age [3]. Dementia (94.7%), muscle weakness (27%), sensory disturbances (28.6%), autonomic nervous dysfunction, ataxia, epilepsy, paroxysmal disturbance of consciousness (39.5%) and parkinsonism are frequently observed in adult-onset NIID patients [4]. While adult-onset NIID is characterized by slowly progressing dementia, it may also present with acute symptoms including stroke-like episodes and epileptic seizures [5]. Dementia and limb weakness are the initial and main clinical manifestations of adult-onset NIID. In most NIID patients over 40 years old, dementia is the first symptom [6]. Diagnosis of the disease formerly required an autopsy but can now be established by skin biopsy and genetic examination [7].
We report a case of NIID with multiple reversible encephalitic attacks, in which the patient experienced two encephalitic episodes followed by complete remission of symptoms. As illustrated by this case, clinicians need to be alert to the possibility of NIID when patients have reversible encephalitic attacks.
Case presentation
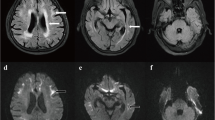
A 63-year-old woman was admitted to hospital with an acute neurological episode. The patient’s complaints were light-headedness, vomiting, unstable gait and cognitive impairment. She had been healthy except for a similar attack seven years prior and had no family history of similar symptoms. Upon admission, the patient’s body temperature was 36.7 °C, and her blood pressure was 122/86 mmHg. Neurological examination revealed no abnormalities in cranial nerve function or muscle strength. The bilateral knee reflex and Achilles tendon reflex were diminished. The biceps and triceps reflexes were normal. The bilateral plantar responses were flexor. Her gait was wide-based and ataxic. She did not have increased muscle tone or bradykinesia. Her complete blood count was normal, as were her liver and kidney function. Neuropsychological examinations revealed mild cognitive impairment: 24/30 on the Mini-Mental State Examination (MMSE), 25/30 on the Montreal Cognitive Assessment (MoCA), 10 on the Hamilton Depression Scale (HAMD), 24 on the Hamilton Anxiety Scale (HAMA), 3 on the Neuropsychiatric Inventory (NPI) and 33 on the Scales for Outcomes in Parkinson’s Disease–Autonomic (SCOPA-AUT). The decreases in the MMSE and MoCA scores were caused mainly by reductions in the patient’s short-term memory and computational ability. Brain MRI revealed high signal intensity in the bilateral frontal grey matter–white matter junction on DWI (Fig. 1 c, d). Cerebrospinal fluid examination revealed mildly elevated levels of protein (0.55 g/l) and glucose (4.5 mmol/l), an absence of red blood cells (0 × 109/l), and a normal leukocyte count (3 × 106 monocytes/l). The patient’s nerve conduction studies were normal. An electroencephalogram (EEG) showed no epileptic discharge. A review of the patient’s brain MRI results from 2011 revealed the presence of a high signal intensity on DWI of the bilateral frontal grey matter–white matter junction (Fig. 1 a, b). According to a detailed inquiry into the patient’s medical history when she was 56 years old, she had experienced light-headedness, right lower quadrant facial droop, unstable gait, aphasia, nausea, vomiting and loss of consciousness. She was admitted to another hospital, where she regained consciousness within 12 h of symptom onset. She received symptomatic treatment, and all other symptoms were completely resolved within one week. The MRI scan showed disease progression compared to the imaging from seven years earlier. Skin biopsy revealed intranuclear inclusions in adipocytes, fibroblasts and sweat gland cells (Fig. 1 e, f, g), which was compatible with NIID. Genetic examination using GC-rich PCR (GC-PCR) and repeat-primed PCR (RP-PCR) found that the patient had 96 GGC repeats in the 5′ UTR of NOTCH2NLC (Fig. 1 h, i). She was not a carrier of the fragile X mental retardation 1 (FMR1) premutation.

Brain magnetic resonance imaging demonstrated high-intensity areas in the cerebral white matter on diffusion-weighted imaging seven years before the most recent hospitalization (a, b). Brain magnetic resonance imaging revealed high-intensity areas in the cerebral white matter and the grey matter–white matter junction on diffusion-weighted imaging (c, d). Skin biopsy samples were subjected to immunohistochemical staining. There were some circular or approximately circular structures deeply stained with 4-dimethylaminoazobenzene in the membranes of sweat gland cells (e: haematoxylin–eosin staining, × 200; f: anti-p62 immunohistochemical staining, × 200; g: electron micrograph). This patient had 96 GGC repeats in NOTCH2NLC (electropherograms of the GC-rich PCR assay [h] and the repeat-primed PCR assay [i])
During hospitalization, the patient was given symptomatic treatment such as anti-dizziness and anti-vomiting treatment as well as supportive treatment such as nutrition support therapy and vitamin B and C supplements during hospitalization, and her symptoms were completely relieved within one week.
In this case study, the patient had two acute encephalitic attacks that were manifested mainly as loss of consciousness, neuropsychological impairments, gait disorders, autonomic dysfunction, etc. Normal body temperature, complete blood count, and cerebrospinal fluid cell counts helped exclude infectious encephalitis. The characteristic high signal intensity on DWI alerted the physicians to perform skin biopsy and genetic testing. The skin biopsy revealed intranuclear inclusions in adipocytes, fibroblasts and sweat gland cells; this set of features was compatible with NIID. A negative test result for the FMR1 gene premutation excluded fragile X–associated tremor ataxia syndrome (FXTAS). The expansion of repeats in the 5′ UTR of the NOTCH2NLC gene confirmed the diagnosis of NIID. At the time of this report, the patient’s outcome is good.
Discussion and conclusions
This case study describes a patient with NIID who presented with two acute reversible encephalitic episodes over seven years. The patient was diagnosed on the basis of characteristic DWI signals, intranuclear inclusions, a negative FMR1 gene premutation and expansion of repeats in the NOTCH2NLC gene.
In light of the patient’s GGC repeat expansion within human-specific NOTCH2NLC and the absence of FMR1 gene premutation, she was diagnosed with sporadic adult-onset NIID [2]. FXTAS is a late-onset neurodegenerative disorder that is similar to NIID in terms of clinical and neuropathological features [8]. We excluded the diagnosis of FXTAS through FMR1 genetic testing, which showed that the number of CGG repeats was within the normal range (< 44 repeats).
Over the seven-year period of disease progression, high DWI signal spread along the grey matter–white matter junction, but the deep white matter was not affected. The signal abnormality progressed continuously once it appeared, which is consistent with previous reports [9].
Due to the highly variable nature of the clinical symptoms, NIID is considered a heterogeneous disease [10]. Most of the previously reported cases showed a degenerative disease course or presented as encephalitis that subsequently deteriorated. One previous case reported a reduced level of consciousness and recurrent vomiting, though the patient’s symptoms continued to progress, which differs from our case [11]. Although multiple reversible encephalitic attacks have been described in case reports previously, some symptoms such as dysarthria recovered completely afterwards, but there were some residual symptoms such as impaired memory and word recall, saccadic pursuit eye movements and mild weakness of the left lower extremity [12,13,14]. In this case, we report a patient with NIID who presented with recurrent loss of consciousness and cognitive impairment that completely recovered within a short time. An important finding was the lack of residual symptoms from the paroxysmal encephalitic episodes. Clinicians should be aware of these findings, and the possibility of NIID should be considered when these symptoms are present. To the best of our knowledge, NIID with multiple reversible attacks has not previously been reported.
NIID is characterized pathologically by the presence of eosinophilic intranuclear inclusions in neuronal and glial cells [3, 15]. Factors contributing to the development of NIID can include abnormal protein accumulation and dysfunction of the intranuclear protein degradation system. The nuclear inclusions (NIs) in the biopsy specimens are immunopositive for ubiquitin and p62 [16]. p62 is an adaptor molecule for the selective autophagic degradation of ubiquitinated proteins [17]. According to recent research, p62 can shuttle into the nucleoplasm and plays an important role in the degradation of intranuclear proteins [18]. It remains unclear what role p62 may play in the pathogenesis of NIID. The inclusions do not seem to cause cell death, but they can disrupt neuronal function [19]. In a study by Nakano [20], ubiquitin- and p62-positive intranuclear aggregates appeared in both controls and NIID cases. However, activated nuclear bodies (NBs) have never been observed in NIID cases, which suggests that the p62- and PML-positive structures might differ in ultrastructural terms between normal and NIID cases. The aberrant response of NBs to stress might lead to fluctuations in clinical symptoms [21]. Accordingly, the dysfunction of NBs or abnormal proteins could be related to the pathogenesis of NIID.
Recently, several studies suggested that NIID was associated with NOTCH2NLC gene alterations [7, 22]. NOTCH2NLC is highly expressed in various populations of radial glia and is associated with evolutionary expansion of the human cerebral cortex [23]. NOTCH2NLC lacks the N-terminal signal peptide and has a 2-bp deletion just downstream of the NOTCH2 start codon. Moreover, by long-read sequencing, GGC repeat expansion in the 5′ region of NOTCH2NLC was identified in all patients with familial NIID as well as those with sporadic NIID. Sone et al. found that the change in the number of repeats ranged from 6 to 61 in control subjects and 71 to 183 in NIID patients [22]. In a study by Tian and colleagues, the number of GGC repeat expansions was less than 40 in healthy subjects and 66 to 517 in NIID patients. Additionally, GGC repeat expansions have been identified as the genetic cause of NIID pathogenesis in the Chinese population [2]. Abnormal GGC duplication in the 5′ UTR region of the human-specific NOTCH2NLC gene is the main cause of NIID and related diseases [7]. GGC repeat expansions may cause neuronal toxicity that leads to NIID [22].
In summary, we present a case of NIID with multiple reversible encephalitic attacks; such a case has never been previously reported. The case was diagnosed by intranuclear inclusions on skin biopsy and by genetic testing; these diagnostics were carried out in response to high-intensity DWI signal at the grey matter–white matter junction. Clinicians need to be alert to the possibility of NIID when patients exhibit reversible encephalitic attacks and intense DWI signal at the grey matter–white matter junction.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Takahashi-Fujigasaki J. Neuronal intranuclear hyaline inclusion disease [J]. Neuropathology. 2003;23(4):351–9.
Deng J, Gu M, Miao Y, et al. Long-read sequencing identified repeat expansions in the 5'UTR of the gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet. 2019;56:758–64. https://doi.org/10.1136/jmedgenet-2019-106268.
Sone J, Kitagawa N, Sugawara E, et al. Neuronal intranuclear inclusion disease cases with leukoencephalopathy diagnosed via skin biopsy. J Neurol Neurosurg Psychiatry. 2014;85:354–6.
Takahashi-Fujigasaki J, Nakano Y, Uchino A, et al. Adult-onset neuronal intranuclear hyaline inclusion disease is not rare in older adults [J]. Geriatr Gerontol Int. 2016;16:51–6.
Chen L, Wu L, Li S, et al. A long time radiological follow-up of neuronal intranuclear inclusion disease: two case reports. Medicine (Baltimore). 2018;97:e13544. https://doi.org/10.1097/MD.0000000000013544.
Cupidi C, Dijkstra AA, Melhem S, et al. Refining the Spectrum of neuronal Intranuclear inclusion disease: a case report. J Neuropathol Exp Neurol. 2019;78:665–70. https://doi.org/10.1093/jnen/nlz043.
Tian Y, Wang JL, Huang W, Zeng S, Jiao B, Liu Z, Chen Z, Li Y, Wang Y, Min HX, et al. Expansion of human-specific GGC repeat in neuronal Intranuclear inclusion disease-related disorders. Am J Hum Genet. 2019;105:166–76.
Greco CM, Berman RF, Martin RM, et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS)[J]. Brain. 2006;129(Pt 1):243–55.
Kawarabayashi T, Nakamura T, Seino Y, et al. Disappearance of MRI imaging signals in a patient with neuronal intranuclear inclusion disease. J Neurol Sci. 2018;388:1–3. https://doi.org/10.1016/j.jns.2018.02.038.
Neuronal intranuclear hyaline inclusion disease. Neuropathology. 2003;23:351–9.
Liu X, Liu X, Du Y, et al. A case of recurrent vomiting: extending the spectrum of neuronal intranuclear inclusion disease [J]. Neurol Sci. 2019;16.
Vermilion J, Johnson M, Srinivasan J. Neuronal Intranuclear inclusion disease: longitudinal case report of motor and nonmotor symptoms. J Child Neurol. 2019;34:801–5. https://doi.org/10.1177/0883073819860566.
Liu Y, Mimuro M, Yoshida M, et al. Inclusion-positive cell types in adult-onset intranuclear inclusion body disease: implications for clinical diagnosis [J]. Acta Neuropathol. 2008;116(6):615–23.
Araki K, Sone J, Fujioka Y, et al. Memory loss and frontal cognitive dysfunction in a patient with adult-onset neuronal Intranuclear inclusion disease [J]. Intern Med. 2016;55(16):2281–4.
Sone J, Tanaka F, Koike H, et al. Skin biopsy is useful for the antemortem diagnosis of neuronal intranuclear inclusion disease [J]. Neurology. 2011;76(16):1372–6.
Sone J , Mori K , Inagaki T , et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease [J]. Brain, 2016:aww249.
Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001;12:2085–90.
Pankiv S, Lamark T, Bruun JA, et al. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem. 2010;285:5941–53.
Afshari H, Kamarei M. Neuronal intranuclear inclusion disease without polyglutamine inclusions in a child.[J]. J Neuropathol Exp Neurol. 2005;64(6):545.
Kimber TE, Blumbergs PC, Rice JP, et al. Familial neuronal intranuclear inclusion disease with ubiquitin positive inclusions [J]. J Neurol Sci. 1998;160(1):33–40.
Nakano Y, Takahashi-Fujigasaki J, Sengoku R, et al. PML nuclear bodies are altered in adult-onset neuronal Intranuclear hyaline inclusion disease [J]. J Neuropathol Exp Neurol. 2017;76(7):585–94.
Sone J, Mitsuhashi S, Fujita A, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease [J]. Nat Genet. 2019;51(8):1215.
Suzuki IK, David G, Roxane VH, et al. Human-Specific, NOTCH2NL, Genes Expand Cortical Neurogenesis through Delta/Notch Regulation [J]. Cell. 2018;173(6):1370–1384.e16.
Acknowledgements
We would like to thank the subject who participated in this study.
Funding
The study received a grant from the Gansu Provincial Health Industry Scientific Research Program (Project number: GSWSKY-2015-57). The funding supported the study design and the collection, analysis, and interpretation of data as well as the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
MML drafted the manuscript; XL collected patient information; YT and LS examined the NOTCH2NLC gene; ZQZ, GDW, WAC, and KL interpreted the data and edited the manuscript. The final manuscript was read and approved by all authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
No competing interests are declared.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, M., Li, K., Li, X. et al. Multiple reversible encephalitic attacks: a rare manifestation of neuronal intranuclear inclusion disease. BMC Neurol 20, 125 (2020). https://doi.org/10.1186/s12883-020-01712-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-020-01712-5