
Abstract
Background
McLeod syndrome is a rare X-linked recessive acanthocytosis associated with neurological manifestations including progressive chorea, cognitive impairment, psychiatric disturbances, seizures, and sensorimotor axonal polyneuropathy. However, no studies have investigated the functioning of central sensorimotor tracts in patients with McLeod syndrome.
Case presentation
A 66-year-old man had experienced slowly progressive chorea and gait disturbance due to lower limb muscle weakness since his early fifties. Blood examinations showed erythrocyte acanthocytosis and the reduction of Kell antigens in red blood cells. Brain magnetic resonance imaging showed atrophy of the bilateral caudate nuclei and putamen. The diagnosis of McLeod syndrome was confirmed by the presence of a mutation of the XK gene on the X chromosome. Somatosensory-evoked potential and transcranial magnetic stimulation studies demonstrated that the central sensory and motor conduction times were abnormally prolonged for the lower extremity but normal for the upper extremity.
Conclusions
This is the first report of the involvement of the central sensorimotor tracts for the legs in a patient with McLeod syndrome. The clinical neurophysiological technique revealed the central sensorimotor tracts involvements clinically masked by neuropathy.
Similar content being viewed by others


Background
McLeod syndrome is an extremely rare progressive X-linked recessive type of neuroacanthocytosis that was first reported by Allen et al. in 1961 [1]. This syndrome is a multisystem disorder with central nervous system (CNS), neuromuscular, cardiovascular, and hematological manifestations. Red blood cell acanthocytosis in McLeod syndrome is associated with absent expression of the Kx antigen and reduced expression of the Kell antigen on the surface membranes of erythrocytes, which are caused by truncation or no expression of XK protein. The detection of mutations in the XK gene confirms a diagnosis of McLeod syndrome. The XK protein plays pivotal roles in organogenesis, cellular structure, and nutrient exchanges [2]. Patients with McLeod syndrome lack expression of this protein, which leads to acanthocytosis and neural degeneration.
Neurological symptoms in McLeod syndrome are various, including progressive chorea, cognitive impairment, psychiatric disturbances, and seizures [3]. Sensorimotor axonal neuropathy is also a typical clinical feature, which leads to distal-dominant muscular weakness with muscular atrophy. A previous pathological study using a mouse model of McLeod syndrome found axonopathy in the spinal cord and the sciatic nerve [4]. However, whether the central sensorimotor tracts are involved in McLeod syndrome remains unclear. For the present study we hypothesized that the central sensorimotor tracts are involved in this disorder.
We used two electrophysiological methods to evaluate the conduction of CNS pathways in McLeod syndrome. First, the central sensory conduction time (CSCT) was measured by recording median and tibial somatosensory-evoked potentials (SEPs). The latencies of the following components were identified: N9 (Erb’s point), N11, N13 (spinal dorsal horn), and N20 (primary sensory cortex) for the median SEP; and N8 (near-field potential of the tibial nerve at the popliteal fossa), N21 (L5–S1 dorsal horn), and P38 (primary sensory cortex) for the tibial SEP (see Table 2 for the montage). The CSCT is calculated as the latency difference between cortical and spinal components. Second, the central motor conduction time (CMCT) was measured using transcranial magnetic stimulation (TMS). TMS can noninvasively elicit motor-evoked potentials (MEPs) by stimulation of the motor cortex or spinal nerve roots; for example, TMS at neural foramina at the C7 and L5 levels elicits MEPs of hand muscles and leg muscles, respectively. The CMCT is defined as the latency difference of MEPs between motor cortical stimulation and spinal root stimulation [5]. Precisely speaking, the CMCT does not purely consist of the corticospinal component, instead including some peripheral component from the nerve root inside the spinal canal. The peripheral component is estimated to be around 0.6 ms for upper-limb muscles, and 1.5 ms or longer for lower-limb muscles since it includes the cauda equina. To overcome the unignorable cauda equina component, we recently reported a new CMCT parameter for the leg muscles named the cortico-conus motor conduction time (CCCT) [6], which is calculated as the MEP latency difference between cortical stimulation and conus stimulation (L1 level). The CCCT can estimate the true central motor conduction without including peripheral components. The new method also allows the cauda equina conduction time (CECT) to be measured, which is defined as the MEP latency difference between stimulation at the L5-level spinal root and L1-level conus [7, 8]. We applied this new TMS method to a patient with McLeod syndrome.
This is the first study to systematically examine the central conduction times in McLeod syndrome. We found significant prolongation of the central conduction for the leg muscles, suggesting that the syndrome involves not only peripheral nerves but also the central sensorimotor tracts.
Case presentations
A 66-year-old man noticed involuntary movements in all extremities and weakness in the lower limb muscles in his early fifties. He had no particular family or past medical history. He was admitted to our hospital with a chief complaint of gait disturbance. On examination, he was conscious and fully oriented, but restless and irritable. He exhibited facial grimacing but no lip or tongue biting. He had chorea in all extremities. He also had right-side-dominant, distal-dominant muscular weakness with muscular atrophy (Medical Research Council Scale grade 1 for the tibialis anterior (TA) muscle and gastrocnemius muscle on the right side and 3 on the left side). His vibratory perception was impaired at the ankles, whereas superficial sensations were intact. He showed a positive Romberg’s sign. Deep tendon reflexes were absent in the extremities, and plantar reflex was indifferent. He needed a cane support in walking.
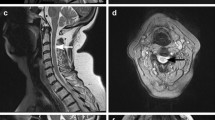
Blood chemical examinations showed elevations of creatine phosphokinase (1609 U/l), aspartate transaminase (54 U/l), alanine transaminase (78 U/l), and lactate dehydrogenase (316 mg/dl). The electrocardiography and chest X-ray findings were normal. The brain magnetic resonance images (MRIs) revealed atrophy of bilateral caudate nuclei and putamen (Fig. 1a), but spinal MRIs showed no abnormalities. Single-photon-emission computed tomography with N-isopropyl-p-[123I]-iodoamphetamine revealed decreases in the blood flow in the basal ganglia.

a MRI fluid-attenuated inversion recovery axial image shows atrophy of the caudate nuclei (arrowheads) and putamen (arrows) bilaterally. b Erythrocyte acanthocytosis present in a peripheral blood smear. c Flow cytometry revealed reduced Kell red blood cell antigens in the peripheral blood (red peak indicates the present case). d Sequencing of the XK gene disclosed mutation c.397C > T (p.Arg133Ter) in exon 2
Table 1 presents the results of the nerve conduction studies. The amplitudes of the compound motor action potentials were reduced in the tibial and fibular nerves, while the amplitudes of the sensory nerve action potentials were reduced in the sural nerves. Needle electromyographic examinations performed at the TA muscle and rectus femoris muscle revealed high-amplitude and long-duration motor unit potentials (MUPs). MUP recruitment was reduced during volitional contraction and fibrillation potentials were present at rest. These findings indicate the presence of chronic denervation and reinnervation processes.
We examined the median and tibial SEPs (Table 2). The right median nerve SEPs showed no delay in any component and no prolongation of CSCT. In the right tibial nerve SEPs, the peripheral components had a normal latency, but the cortical latency was prolonged. CSCT was also abnormally prolonged (19.5 ms; normal < 13.2 ms).
We used TMS to examine MEPs from the right first-dorsal interosseous (FDI) muscle and TA muscle (Table 3). MEP latencies for the FDI were normal for stimulation to the cortex, brainstem, and cervical nerve root. The CMCT was also within the normal range (6.5 ms; normal < 7.7 ms) [9]. MEP could not be evoked from the right TA muscle due to severe muscle atrophy, and so we recorded MEPs from the left TA muscle. MEP latencies were prolonged for stimulation to the cortex and brainstem, but normal for stimulation at levels L1 and L5. Both the conventional CMCT (19.3 ms; normal < 17.0 ms) and the CCCT (15.9 ms; normal < 14.7 ms) were abnormally prolonged [6] (Fig. 2). The CECT was normal.

TMS findings. a Schematic of sites where magnetic stimulation was applied. b MEP latencies for cortical and brainstem stimulation are prolonged, while those for stimulation at levels L1 (conus) and L5 (neuro-foramina) are within the normal ranges. Both the conventional CMCT and the CCCT are abnormally prolonged
The peripheral blood smear disclosed frequent acanthocytes (28% of red cells, Fig. 1b), and flow cytometry disclosed weak expression of Kell red blood cell antigens (Fig. 1c). On the basis of progressive chorea, psychiatric symptoms, sensorimotor axonal neuropathy in the lower extremities, and acanthocytosis, we performed gene sequencing of his blood samples. Sequence analysis of the XK gene detected mutation c.397C > T (p.Arg133Ter) in exon 2 (reference sequence NM_021083.2) (Fig. 1d). According to this gene mutation, he was diagnosed with McLeod syndrome [10].
He continued to take oral haloperidol at a dosage of 1.5 mg/day, and his chorea and restlessness were found to be well controlled in outpatient clinic examinations.
Discussion and conclusions
Our patient had progressive chorea, psychiatric disturbances, distal-dominant muscular weakness with atrophy in the lower extremities, and acanthocytosis, which match the clinical features of McLeod syndrome. The diagnosis was confirmed by detection of a previously known mutation in the XK gene [3, 11, 12]. Nerve conduction studies showed sensorimotor axonal neuropathy only in the lower extremities. The cortical latency of the SEPs was also delayed for tibial nerve stimulation, suggesting the involvement of the central sensory pathway that includes the dorsal cord, medial lemniscus, thalamus, and primary sensory cortex. In addition, the TMS technique revealed the involvement of the corticospinal tract for the leg muscles (prolongation of the CMCT and CCCT). Together these findings indicate the involvement of the sensorimotor tracts in both the CNS and peripheral nervous system, mainly those for the lower extremities.
Muscular atrophy in McLeod syndrome has been explained by motor axonal neuropathy. A muscle biopsy can detect a combination of neurogenic and myogenic changes, with the former being more obvious [3]. Sensory symptoms have also been attributed to sensory axonal neuropathy [11, 13]. However, the pathogenesis of the peripheral neuropathy remains to be determined.
This is the first demonstration of the involvement of the central sensorimotor tracts in McLeod syndrome. The findings of this study suggest the presence of length-dependent axonal degeneration of the sensorimotor tract fibers in McLeod syndrome. This is especially interesting given that a previous pathological study found that model knockout mice of McLeod syndrome showed axonopathy in the spinal cord and the sciatic nerve [4]. The present findings provide clinical proof for these experimental results, which is important for understanding the distribution of this multisystem disorder.
A unique finding of the present case study is that the TMS technique revealed the involvement of the corticospinal tract even in a patient whose plantar responses were indifferent. When both the corticospinal tract and peripheral nerves are damaged, Babinski signs may not be observed, because the peripheral pathology masks the central pathology. TMS can allow evaluation of corticospinal tract function even in this situation [9]. The lack of evidence for corticospinal tract involvement in the previous clinical reports might be due to coexisting peripheral neuropathy, which is common in McLeod syndrome. Similarly, measuring SEPs is also useful for detecting the involvement of central sensory pathways in patients with peripheral neuropathy.
In conclusion, the results obtained in this study suggest that McLeod syndrome involves the central sensorimotor tracts in addition to peripheral nerves. This involvement of central sensorimotor tracts for the legs might be masked by the presence of peripheral neuropathy in this disorder.
Availability of data and materials
Not applicable.
Abbreviations
- CCCT:
-
Cortico-conus motor conduction time
- CECT:
-
Cauda equina conduction time
- CMCT:
-
Central motor conduction time
- CNS:
-
Central nervous system
- CSCT:
-
Central sensory conduction time
- FDI:
-
First-dorsal interosseous
- MEP:
-
Motor-evoked potential
- MRI:
-
Magnetic resonance image
- MUP:
-
Motor unit potential
- NCS:
-
Nerve conduction study
- SEP:
-
Somatosensory-evoked potential
- TA:
-
Tibialis anterior
- TMS:
-
Transcranial magnetic stimulation
References
Allen FH Jr, Krabbe SM, Corcoran PA. A new phenotype (McLeod) in the Kell blood-group system. Vox Sang. 1961;6:555–60.
Roulis E, Hyland C, Flower R, Gassner C, Jung HH, Frey BM. Molecular basis and clinical overview of McLeod syndrome compared with other Neuroacanthocytosis syndromes: a review. JAMA Neurol. 2018;75(12):1554–62.
Danek A, Rubio JP, Rampoldi L, Ho M, Dobson-Stone C, Tison F, Symmans WA, Oechsner M, Kalckreuth W, Watt JM, et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol. 2001;50(6):755–64.
Zhu X, Cho ES, Sha Q, Peng J, Oksov Y, Kam SY, Ho M, Walker RH, Lee S. Giant axon formation in mice lacking Kell, XK, or Kell and XK: animal models of McLeod neuroacanthocytosis syndrome. Am J Pathol. 2014;184(3):800–7.
Rossini PM, Burke D, Chen R, Cohen LG, Daskalakis Z, Di Iorio R, Di Lazzaro V, Ferreri F, Fitzgerald PB, George MS, et al. Non-invasive electrical and magnetic stimulation of the brain, spinal cord, roots and peripheral nerves: basic principles and procedures for routine clinical and research application. An updated report from an I.F.C.N. committee. Clin Neurophysiol. 2015;126(6):1071–107.
Matsumoto H, Hanajima R, Shirota Y, Hamada M, Terao Y, Ohminami S, Furubayashi T, Nakatani-Enomoto S, Ugawa Y. Cortico-conus motor conduction time (CCCT) for leg muscles. Clin Neurophysiol. 2010;121(11):1930–3.
Matsumoto H, Hanajima R, Terao Y, Hamada M, Shirota Y, Yugeta A, Nakatani-Enomoto S, Hashida H, Ugawa Y. A significant correlation between cauda equina conduction time and cerebrospinal fluid protein in chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Sci. 2018;384:7–9.
Matsumoto H, Hanajima R, Terao Y, Yugeta A, Hamada M, Shirota Y, Ohminami S, Nakatani-Enomoto S, Tsuji S, Ugawa Y. Prominent cauda equina involvement in patients with chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Sci. 2010;290(1–2):112–4.
Ugawa Y, Uesaka Y, Terao Y, Suzuki M, Sakai K, Hanajima R, Kanazawa I. Clinical utility of magnetic corticospinal tract stimulation at the foramen magnum level. Electroencephalogr Clin Neurophysiol. 1996;101(3):247–54.
Urata Y, Nakamura M, Sasaki N, Shiokawa N, Nishida Y, Arai K, Hiwatashi H, Yokoyama I, Narumi S, Terayama Y, et al. Novel pathogenic XK mutations in McLeod syndrome and interaction between XK protein and chorein. Neurol Genet. 2019;5(3):e328.
Dotti MT, Battisti C, Malandrini A, Federico A, Rubio JP, Circiarello G, Monaco AP. McLeod syndrome and neuroacanthocytosis with a novel mutation in the XK gene. Mov Disord. 2000;15(6):1282–4.
Bansal I, Jeon HR, Hui SR, Calhoun BW, Manning DW, Kelly TJ, Lee S, Baron BW. Transfusion support for a patient with McLeod phenotype without chronic granulomatous disease and with antibodies to Kx and km. Vox Sang. 2008;94(3):216–20.
Malandrini A, Fabrizi GM, Truschi F, Di Pietro G, Moschini F, Bartalucci P, Berti G, Salvadori C, Bucalossi A, Guazzi G. Atypical McLeod syndrome manifested as X-linked chorea-acanthocytosis, neuromyopathy and dilated cardiomyopathy: report of a family. J Neurol Sci. 1994;124(1):89–94.
Acknowledgments
The authors thank Ms. Yumiko Tanji for assistance in the electrophysiological examinations.
Funding
Dr. Murakami has received research funding from the Takeda Science Foundation. Dr. Ugawa was partly supported by research grants from JSPS KAKENHI Grant Number 15H05881, 16H05322, and 25293206.
Author information
Authors and Affiliations
Contributions
TM managed the patient, conducted electrophysiological evaluations and wrote the first draft of the manuscript. DA, RT, MA, AT, SK and CK managed the patient and revised the manuscript. HM performed electrophysiological evaluations and revised the manuscript. YUr, MN and AS performed gene analyses and revised the manuscript. YUg revised the manuscript and provided intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient and his wife for the publication of this case report and any accompanying images. A copy of the written consent is available for review by the editor of this journal.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Murakami, T., Abe, D., Matsumoto, H. et al. A patient with McLeod syndrome showing involvement of the central sensorimotor tracts for the legs. BMC Neurol 19, 301 (2019). https://doi.org/10.1186/s12883-019-1526-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-019-1526-9