
Abstract
Background
Primary coenzyme Q10 (CoQ10) deficiency of genetic origin is one of a few treatable focal segmental glomerulosclerosis (FSGS). Renal morphologic evidence for COQ8B mutation and CoQ10 deficiencies of other gene mutations is assessed using electron microscopy with marked increase of abnormal-shaped mitochondria in podocytes. However, light microscopic morphologic features of deficiencies other than FSGS have not been reported.
Case presentation
A 30-year-old woman was admitted to our hospital because proteinuria was found during four consecutive medical checkups. She had no medical history or family history of proteinuria and severe renal dysfunction. The swollen podocytes were stained to the same extent as mitochondria-rich proximal tubular cells under both Masson’s trichrome and hematoxylin-eosin staining, whereas no mitochondrial abnormalities were detected under the first electron microscopic views. As proteinuria and estimated glomerular filtration rate (eGFR) deteriorated after pregnancy, we reevaluated the additional electron microscopic views and detected mitochondrial abnormalities. Genetic testing revealed COQ8B mutation (c.532C > T, p.R178W); therefore, we diagnosed COQ8B nephropathy. CoQ10 supplementation improved proteinuria and stopped eGFR reduction.
Conclusions
This is the first report of granular swollen podocytes due to mitochondrial diseases detected under light microscopy. We propose that this finding can be the clue for the diagnosis of both COQ8B nephropathy and the other CoQ10 deficiencies.
Similar content being viewed by others


Background
Focal segmental glomerulosclerosis (FSGS) is a leading cause of end-stage kidney disease in children and adults [1]. Steroid-resistant nephrotic syndrome (SRNS) is a glomerular disease characterized by massive proteinuria, most often associated with FSGS. Among almost 50 genes identified as causes of FSGS and/or SRNS [2,3,4], primary coenzyme Q10 (CoQ10) deficiency of genetic origin is one of a few treatable FSGS and/or SRNS [4,5,6,7,8,9,10,11,12]. CoQ10 is a mitochondrial coenzyme that is essential for the mitochondrial respiratory chain and ATP production. A set of at least 17 different genes synthesized CoQ10 in mitochondria [13,14,15,16,17]. Of these, mutations in ten genes (prenyl diphosphate synthase subunit 1 [PDSS1], PDSS2, COQ2, COQ4, COQ6, COQ7, COQ8A, COQ8B, COQ9, and AarF domain containing kinase 2) cause primary CoQ10 deficiency [17, 18]. Many signs and symptoms of CoQ10 deficient patients are common among other mitochondrial diseases; these involve multiple organ systems and often show prominent neurologic and myopathic features. However, glomerular dysfunction, such as early-onset FSGS and/or SRNS, is peculiar to CoQ10 deficiencies because of the following mutations: PDSS2 [6, 7], COQ2 [8], COQ6 [9, 10], and COQ8B [11, 12, 19,20,21,22,23,24]. Of four genes, mutation in COQ8B causes selective glomerular phenotype mostly without neurological and myopathic deficits [11, 12, 19,20,21,22,23,24]. The age at onset is usually between 5 and 20 years [11, 12, 19,20,21,22,23,24,25].
Renal morphologic evidence for COQ8B mutation and CoQ10 deficiencies of the other gene mutations is a marked increase of abnormal-shaped mitochondria in podocytes as assessed using electron microscopy [8, 10, 20, 21]. In the other mitochondria diseases, the accumulation of abnormal mitochondria is more likely seen in tubular cells than podocytes under electron microscope because mitochondriopathies typically cause tubulopathy [26]. Consistent with this, granular swollen epithelial cells (GSECs) in tubular cells are easily detected under light microscope [27]; these cells are a distinct morphologic feature that suggests mitochondrial diseases. However, swollen podocytes accumulating abnormal mitochondria remain undetected by light microscope. Additionally, light microscopic morphologic features other than FSGS under CoQ10 deficiencies are also lacking. Sampling error for electron microscopic material may greatly affect the finding of sporadic abnormal podocytes; therefore, identifying abnormal podocytes by light microscopy may be helpful for suspecting COQ8B nephropathy and other CoQ10 deficiencies. Here, we report the case of a patient with homozygous COQ8B mutation (c.532C > T, p.R178W) who presented an adult-onset FSGS without family history. Light microscopy clearly showed granular swollen podocytes, which corresponded with podocytes filled with numerous dysmorphic mitochondria by electronic microscopy.
Case presentation
A 30-year-old Japanese woman with no chief complaints was admitted to our hospital because proteinuria was found during four consecutive medical checkups. Urinary protein level was assessed at her medical checkup every year. She had no medical history and family history of proteinuria or severe renal dysfunction. On admission, the physical examination did not show notable abnormalities; her blood pressure was normal (110/64 mmHg) and no edema was observed. The initial laboratory evaluation (Table 1) showed a normal complete blood count; serum creatinine, 0.64 mg/dL; estimated glomerular filtration rate (eGFR), 82 mL/min per 1.73 m2; total protein 7.3 g/dL; albumin 4.5 g/dL. Urinalysis revealed 2+ proteinuria; urine protein creatinine ratio (uPCR) was 1.85 g/gCr. Serologic workup results were negative. Ultrasonography of the kidneys showed that they were of normal size.
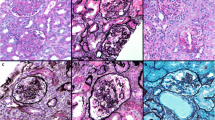
In the first renal biopsy, 31 glomeruli, including 13 global (Fig. 1a) and 2 segmental scleroses (Fig. 1b and c), were obtained. In addition to podocyte detachment, the swollen podocytes were red-stained to the same extent as mitochondria-rich proximal tubular cells in the segmental sclerotic glomeruli under both Masson’s trichrome and hematoxylin-eosin (HE) staining (Fig. 1b and c), but the remaining glomeruli were almost normal and no significant deposition was observed in immunofluorescence. Foot processes were segmentally effaced under electron microscope, but no mitochondrial abnormalities were detected. As she did not want to examine the cause of FSGS, which had not been otherwise specified by genetic testing at this point, angiotensin receptor blocker (ARB) was started as a symptomatic treatment. This decreased her urinary protein excretion during the almost-1-year follow-up (Fig. 2). However, after ARB was discontinued for pregnancy, her uPCR increased and nephrotic syndrome developed with proteinuria, hypoalbuminemia, and decreased eGFR developed (Fig. 2). To detect mitochondrial abnormalities, we reevaluated the additional electron microscopic views, which revealed that some podocyte cell bodies were filled with numerous dysmorphic mitochondria lacking cristae or with abnormally enlarged ones (Fig. 1d). Accordingly, podocytes were also characterized by extensive foot process effacement and marked hypertrophy (Fig. 1d). Because mitochondrial abnormalities in podocytes were suspected, we analyzed genomic DNA from her peripheral blood mononuclear cells by targeted resequencing using next-generation sequencing, which revealed the existence of homozygous missense mutations c.532C > T (p.R178W) in exon 6 of COQ8B gene, which led to a definitive diagnosis of mitochondrial disease.

Light and electron microscopic findings at first (a-d) and second biopsy (e-g). a, e Periodic acid–Schiff (PAS) staining at a low magnification (×100) in the first and second biopsy, respectively. Global or segmental sclerotic glomeruli (indicated by arrow) were increased at the second biopsy (e). Scale bars, 100 μm. b, c Segmental sclerotic glomerular images of Masson’s trichrome (b) and hematoxylin-eosin (HE) staining (c) at a high magnification (× 400). Granular swollen podocytes were indicated by arrowheads. Scale bars: 20 μm. d Electron microscopic views of podocytes filled with abnormal mitochondria were observed (indicated by arrowheads). Segmental foot process effacement of the epithelial cells was observed (lower left panel). Numerous dysmorphic mitochondria lacking cristae or with abnormally enlarged ones were observed in podocyte cell bodies (lower right panel). Scale bars: 5 μm. f Light microscopic views of the granular swollen podocytes (indicated by arrowheads) in the same focal segmental glomerulus under Masson’s trichrome (upper left panels), HE (upper right panels), PAS (lower left panels), and Periodic acid–methenamine-silver staining (lower right panels) (× 400). Scale bars: 20 μm. g Granular swollen podocytes (indicated by white arrowheads) were observed in sclerotic glomeruli (upper panels), but not normal-appearing glomeruli (lower panels) under Masson’s trichrome staining (× 400). Normal podocytes are indicated by black arrowheads. Scale bars: 20 μm. h Immunochemistry showing increased segmental staining of COX IV in glomeruli from this patient (left panel), but not in those from the control patient with no mitochondrial disease (right panel). COX IV-positive podocytes and normal podocytes are indicated by white and black arrowheads, respectively. Scale bars: 20 μm

Patient’s clinical course. Changes in the estimated glomerular filtration rate (GFR, green lines), serum albumin (Alb, orange lines), urine protein/creatinine ratio (uPCR, blue bars). CoQ10: coenzyme Q10, ARB: angiotensin receptor blocker
Before the CoQ10 supplementation, we performed a follow-up renal biopsy. In the second renal biopsy, 31 glomeruli were obtained, including 22 global (Fig. 1e) and 3 segmental scleroses, which shows the exacerbation of the percentage of glomerulosclerosis and tubulointerstitial fibrosis. As shown in Fig. 1f, Masson’s trichrome staining clearly showed granular swollen podocytes, consistent with the findings of the first biopsy. These podocytes were distinguished from protein droplets in cases with proteinuria because the absorptive protein droplets are positive for Periodic acid–methenamine-silver (PAM) staining [23]. Ten of these podocytes were observed among six glomeruli with scleroses and/or adhesion, but not among normal-appearing glomeruli (Fig. 1g). In both the first and second biopsies, no dysmorphic mitochondria were detected in other glomerular cells, including parietal, endothelial, and mesangial cells, or in myocytes of small arteries, interstitial fibroblasts, and tubular epithelial cells. After this second renal biopsy, she received treatment with oral CoQ10 at a dose of 10–20 mg/kg/day. This supplementation decreased uPCR and stopped eGFR reduction (Fig. 2).
Discussion and conclusions
We reported the case of adult-onset COQ8B nephropathy without family history of proteinuria or sever renal dysfunction presenting with FSGS. Additionally, we showed that granular swollen podocytes were identified under light microscope and that Masson trichrome and HE staining clearly showed them. In these podocytes, protein droplets were negative for PAM stain, which suggests that they include mitochondria to the same extent as mitochondria-rich proximal tubular cells. Although the kinds of cell with mitochondrial accumulation differ, Kobayashi et al. have found GSECs among distal tubules and collecting ducts in all patients with mutated mtDNA A3243G and T3271C, and have also demonstrated that Masson trichrome stain is the best way to detect these cells because it stains mitochondria red [27]. Therefore, our light microscopic findings of podocytes due to COQ8B nephropathy appear to be similar to those of GSECs in tubules due to mitochondrial diseases [27], which suggests that this is the first report to detect GSECs in podocytes due to mitochondrial diseases under light microscopy. Moreover, increased podocyte expression of a mitochondrial-specific protein COX IV in this patient supports the observation that light microscopy can detect accumulation of mitochondria in podocytes (Fig. 1h). It might be difficult to clearly detect red-stained granules in all abnormal podocytes because their cells are smaller than tubular cells. However, under CoQ10 deficiencies, we considered GSECs in podocytes to be more important than those in tubules because CoQ10 deficiencies are more likely to cause glomerular dysfunction than tubular injury [11, 12, 19,20,21], as opposed to the other mitochondrial diseases [26]. Additionally, as COQ8B nephropathy may progress much more quickly than other genetic FSGS, such as NPHS2 and WT1 nephropathy [12], it is important to early suspect COQ8B nephropathy and properly diagnose. In our case, proteinuria rapidly increased during pregnancy and reached a nephrotic level although the deterioration might be affected by ARB discontinuation and pregnancy itself. Therefore, GSECs in podocytes appear to be a useful tool to suspect CoQ8B nephropathy and prevent their exacerbation by early diagnosis.
We observed more GSECs in podocytes in the second biopsy than the first. Both urinary protein and serum creatinine levels in the second biopsy were higher than the first, which was consistent with exacerbation of percent glomerulosclerosis and tubulointerstitial fibrosis. However, GSECs in tubular cells are uncorrelated with these parameters [27]. Interestingly, we observed GSECs in podocytes among only glomeruli with scleroses and/or adhesion, but not among normal-appearing glomeruli. This specific localization speculates that GSECs in podocytes may cause FSGS through podocytopathy. Although COQ8B knockdown in cultured podocytes does not affect apoptosis, loss of COQ8B reduces podocyte migration [11]. Both this impaired podocyte migration in vitro and proteinuria in clinical conditions are reversed by CoQ10 addition [11, 12, 21,22,23,24]. Consistent with these reports, treatment with CoQ10 decreased urinary protein level of our patient. Therefore, we speculate that GSECs in podocytes lead to glomerulosclerosis through podocytopathy and may be associated with both clinical and histological parameter of kidney function.
This patient had no neurological or myopathic features, which are common in mitochondrial diseases. Consistent with these observations, COQ8B mutations cause selective glomerular phenotypes, mostly without neurological and myopathic deficits [11, 12, 19,20,21,22,23,24]. Although the mechanism of this selectivity remains unclear, a potential explanation may be the difference in distribution patterns between COQ8B and COQ8A with high sequence similarity [11, 12]. COQ8B is highly expressed in podocytes, whereas COQ8A is expressed in most body tissues but not podocytes [11]. Neurologic abnormalities were reported in only 14–24% of patients with COQ8B nephropathy [12, 19]. Although we cannot completely exclude the possibility of extrarenal involvement, the patient was clinically considered to have presented a selective glomerular phenotype.
To date, 89 patients have been reported to have been diagnosed with COQ8B nephropathy and genetic analysis revealed 30 COQ8B mutations [11, 12, 19,20,21,22,23,24,25]. In this case, we found COQ8B mutation (c.532C > T, p.R178W) using genetic analysis. Although this mutation is a known pathogenic missense mutation [11, 12], we considered this case to be unique because the family history was negative for late-onset COQ8B nephropathy (Fig. 3). In cases with positive family history, 22.2% of patients developed COQ8B nephropathy aged 20 or older, but there were no reports of adult-onset COQ8B nephropathy in cases with negative family history (Fig. 3) [11, 12, 19,20,21,22,23,24,25]. One possible explanation for this difference in onset is that underdiagnosis is more likely in cases with negative family history because the locations of mutations are not strongly correlated with age at onset. In fact, frameshift mutations of c.1447G > T completely abolish COQ8B function in vitro [13], but similar frameshift homozygous mutations, such as 1199dupA, 1339dupG, and 1356_1362delGGGCCCT, do not always cause early onset (Fig. 3). Although mitochondrial diseases are diagnosed using genetic analysis or electron microscopy when it is suspected, genetic testing can be performed at only specific facilities. Thus, just as in our case, it is difficult to diagnose COQ8B nephropathy when podocytes accumulating abnormal mitochondria are not included in electron microscopic views. Therefore, it is important to carefully suspect CoQ10 deficiencies in cases in which patients with proteinuria, even if they are aged over 20 years old and lack a family history of proteinuria and severe renal dysfunction.

Age at onset of COQ8B nephropathy with negative or positive family history. The data were obtained from the previous reports [11, 12, 19,20,21,22,23,24,25] and negative or positive family histories were separately plotted as open or closed circles, respectively. This case is indicated by red open circles. Heterozygous or homozygous mutations are indicated as normal or bold letters, respectively
We cannot refer to the sensitivity or specificity of these podocyte findings because it is extremely difficult to obtain sufficient numbers of patients for rare diseases. Because Masson’s trichrome staining was not demonstrated in previous report of COQ8B nephropathy [11, 12, 19,20,21,22,23,24], further studies may be necessary to carefully evaluate and determine the scientific relevance of this staining.
In conclusion, we reported the case of patient with COQ8B mutation (c.532C > T, p.R178W) who presented with granular swollen podocytes in segmental and global sclerotic glomeruli under light microscope. When GSECs are found in podocytes, suspecting mitochondrial disease may lead to a faster and more accurate diagnosis. Therefore, we propose that this finding can be the clue for the diagnosis of mitochondrial nephropathy. As few patients develop COQ8B nephropathy aged 20 or older, it is important to carefully suspect CoQ10 deficiencies. Further studies using more specimens are needed to confirm our findings.
Availability of data and materials
The datasets during this case report are available from the corresponding author on reasonable request.
Abbreviations
- COQ:
-
coenzyme Q
- FSGS:
-
focal segmental glomerulosclerosis
- SRNS:
-
steroid-resistant nephrotic syndrome
- PDSS:
-
prenyl diphosphate synthase subunit
- GSECs:
-
granular swollen epithelial cells
- eGFR:
-
estimated glomerular filtration rate
- uPCR:
-
urine protein/creatinine ratio
- HE:
-
hematoxylin-eosin
- ARB:
-
angiotensin receptor blocker
- PAM:
-
periodic acid–methenamine-silver
References
D'Agati V. Pathologic classification of focal segmental glomerulosclerosis. Semin Nephrol. 2003;23(2):117–34.
Bierzynska A, Soderquest K, Koziell A. Genes and podocytes - new insights into mechanisms of podocytopathy. Front Endocrinol (Lausanne). 2014;5:226.
Lovric S, Fang H, Vega-Warner V, Sadowski CE, Gee HY, Halbritter J, Ashraf S, Saisawat P, Soliman NA, Kari JA, et al. Rapid detection of monogenic causes of childhood-onset steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2014;9(6):1109–16.
Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26(6):1279–89.
DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348(26):2656–68.
López LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79(6):1125–9.
Peng M, Falk MJ, Haase VH, King R, Polyak E, Selak M, Yudkoff M, Hancock WW, Meade R, Saiki R, et al. Primary coenzyme Q deficiency in Pdss2 mutant mice causes isolated renal disease. PLoS Genet. 2008;4(4):e1000061.
Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer L, Barisoni L, Pastore A, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol. 2007;18(10):2773–80.
Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z, Xie LX, Salviati L, Hurd TW, Vega-Warner V, et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest. 2011;121(5):2013–24.
Widmeier E, Airik M, Hugo H, Schapiro D, Wedel J, Ghosh CC, Nakayama M, Schneider R, Awad AM, Nag A, et al. Treatment with 2,4-Dihydroxybenzoic acid prevents FSGS progression and renal fibrosis in Podocyte-specific. J Am Soc Nephrol. 2019;30:393–405.
Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, Fang H, Song X, Cattran DC, Avila-Casado C, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123(12):5179–89.
Korkmaz E, Lipska-Ziętkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, Schnaidt S, Gucer S, Kaymaz F, Arici M, et al. ADCK4-associated Glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol. 2016;27(1):63–8.
Fonseca LV, Doimo M, Calderan C, Desbats MA, Acosta MJ, Cerqua C, Cassina M, Ashraf S, Hildebrandt F, Sartori G, et al. Mutations in COQ8B (ADCK4) found in patients with steroid-resistant nephrotic syndrome alter COQ8B function. Hum Mutat. 2018;39(3):406–14.
Doimo M, Desbats MA, Cerqua C, Cassina M, Trevisson E, Salviati L. Genetics of coenzyme q10 deficiency. Mol Syndromol. 2014;5(3–4):156–62.
Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J Inherit Metab Dis. 2015;38(1):145–56.
Payet LA, Leroux M, Willison JC, Kihara A, Pelosi L, Pierrel F. Mechanistic details of early steps in coenzyme Q biosynthesis pathway in yeast. Cell Chem Biol. 2016;23(10):1241–50.
Vázquez-Fonseca L, Schaefer J, Navas-Enamorado I, Santos-Ocaña C, Hernández-Camacho JD, Guerra I, Cascajo MV, Sánchez-Cuesta A, Horvath Z, Siendones E, et al. ADCK2 Haploinsufficiency Reduces Mitochondrial Lipid Oxidation and Causes Myopathy Associated with CoQ Deficiency. J Clin Med. 2019;8(9):1374.
Acosta MJ, Vazquez Fonseca L, Desbats MA, Cerqua C, Zordan R, Trevisson E, Salviati L. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta. 2016;1857(8):1079–85.
Wang F, Zhang Y, Mao J, Yu Z, Yi Z, Yu L, Sun J, Wei X, Ding F, Zhang H, et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr Nephrol. 2017;32(7):1181–92.
Park E, Kang HG, Choi YH, Lee KB, Moon KC, Jeong HJ, Nagata M, Cheong HI. Focal segmental glomerulosclerosis and medullary nephrocalcinosis in children with ADCK4 mutations. Pediatr Nephrol. 2017;32(9):1547–54.
Yang J, Yang Y, Hu Z. A novel ADCK4 mutation in a Chinese family with ADCK4-associated glomerulopathy. Biochem Biophys Res Commun. 2018;506(3):444–9.
Lolin K, Chiodini BD, Hennaut E, Adams B, Dahan K, Ismaili K. Early-onset of ADCK4 glomerulopathy with renal failure: a case report. BMC Med Genet. 2017;18(1):28.
Atmaca M, Gulhan B, Korkmaz E, Inozu M, Soylemezoglu O, Candan C, Bayazıt AK, Elmacı AM, Parmaksiz G, Duzova A, et al. Follow-up results of patients with ADCK4 mutations and the efficacy of CoQ10 treatment. Pediatr Nephrol. 2017;32(8):1369–75.
Feng C, Wang Q, Wang J, Liu F, Shen H, Fu H, Mao J. Coenzyme Q10 supplementation therapy for 2 children with proteinuria renal disease and ADCK4 mutation: case reports and literature review. Medicine (Baltimore). 2017;96(47):e8880.
Zhang H, Wang F, Liu X, Zhong X, Yao Y, Xiao H. Steroid-resistant nephrotic syndrome caused by co-inheritance of mutations at NPHS1 and ADCK4 genes in two Chinese siblings. Intractable Rare Dis Res. 2017;6(4):299–303.
Chinnery PF. Mitochondrial disorders overview. In: GeneReviews, edited by Pagon RA, Adam MP, Ardinger HH, et al, Seattle, WA, University of Washington, Seattle. http://www.ncbi.nlm.nih.gov/books/NBK1224. Accessed 22 Jan 2020.
Kobayashi A, Goto Y, Nagata M, Yamaguchi Y. Granular swollen epithelial cells: a histologic and diagnostic marker for mitochondrial nephropathy. Am J Surg Pathol. 2010;34(2):262–70.
Acknowledgements
We thank Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript.
Funding
Genetic testing was supported by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED (19ek0109273) to KM, YO and TI (http://www.amed.go.jp/en/).
Author information
Authors and Affiliations
Contributions
TD, KM1 and SH treated the patient and examined renal biopsies. TD, MA, KM1, SH1, YM2, TI, SH2, and TM interpreted pathologic findings. YK, YO, and KM2 examined genetic testing. YM1 drafted the manuscript. TD, YM2, and TM revised the manuscript critically. All authors read and approved the final manuscripts.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee at which the studies were conducted (IRB approval number Hi-204) and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
Consent for publication
Written consent to publish this information was obtained from the patient. A copy of the consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Maeoka, Y., Doi, T., Aizawa, M. et al. A case report of adult-onset COQ8B nephropathy presenting focal segmental glomerulosclerosis with granular swollen podocytes. BMC Nephrol 21, 376 (2020). https://doi.org/10.1186/s12882-020-02040-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-020-02040-z