
Abstract
Background
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary renal disorder, leading to end stage renal failure and kidney transplantation in its most serious form. The severity of the disease’s manifestation depends on the genetic determination of ADPKD. The huge variability of different phenotypes (even within a single family) is not only modulated by the two main ADPKD genes (PKD1 and PKD2) but also by modifier genes and the whole genetic background.
Case presentation
This is a report of an ADPKD family with co-inheritance of PKD1 and PKD2 pathogenic variants. The proband, with an extremely serious manifestation of ADPKD (the man was diagnosed in early childhood, and with end stage renal disease aged 23), underwent genetic analysis of PKD1 and PKD2, which revealed the presence of pathogenic mutations in both of these genes. The missense PKD2 mutation p.Arg420Gly came from the proband’s father, with a mild ADPKD phenotype. The same mutation of the PKD2 gene and similar mild disease presentation were found in the proband’s aunt (father’s sister) and her son. The nonsense mutation p.Gln2196* within the PKD1 gene was probably inherited from the proband’s mother, who died at the age of 45. It was only discovered post mortem, that the real cause of her death was kidney failure as a consequence of untreated ADPKD. Unfortunately, neither the DNA of the proband’s mother nor the DNA of any other family members from this side of the pedigree were available for further examination. The proband underwent successful cadaveric kidney transplantation at the age of 24, and this replacement therapy lasted for the next 15 years.
Conclusions
Here, we present a first case of bilineal ADPKD inheritance in the Czech Republic. This report highlights the significant role of modifier genes in genetic determination of ADPKD, especially in connection with seriously deteriorated disease phenotypes. In our case, the modifying role is probably mediated by the PKD2 gene.
Similar content being viewed by others


Background
Autosomal dominant polycystic kidney disease (ADPKD; OMIM *601313 for PKD1, and *173910 for PKD2) is the most common hereditary kidney disease, predominantly characterized by the presence of cysts in both kidneys leading to end-stage renal disease (ESRD), usually in adulthood [1]. Its incidence is 1 per 400–1000 persons in the general population. The systemic and multi-organ nature of ADPKD manifests by the occurrence of cysts in extrarenal organs (liver, pancreas, spleen), as well as by other manifestations such as cardiovascular abnormalities and/or brain vessel aneurysms [2, 3]. Cystogenesis is associated with the development of arterial hypertension, which precedes the deterioration of the glomerular filtration rate in a notable proportion of ADPKD patients. Alterations in epithelial cell growth, fluid secretion, and extracellular matrix composition are the main pathophysiological abnormalities observed during the onset and growth of the cysts. These processes are caused by the deregulation of cAMP, as well as EGFR- and mTOR-mediated pathways. Two genes (PKD1 and PKD2) have been uncovered as being causative in ADPKD [4, 5]. PKD1 and PKD2 code for two gene products: polycystin 1 (PC-1) and polycystin 2 (PC-2), respectively. The germline mutations in PKD1 are present in approximately 85% of the ADPKD patients (ADPKD type 1); mutations in PKD2 in the remaining 15% of ADPKD patients (ADPKD type 2). However, the portion of PKD2 mutations can rise to 36% in community-based studies [6]. Clinical manifestations due to defective PC-1 in the PKD1 genotype and PC-2 in the PKD2 genotype display a phenotypic similarity, which is largely explained by the interaction between both polycystins. PC-1 and PC-2, together with fibrocystin/polyductin, which is responsible for autosomal recessive polycystic kidney disease (ARPKD), form a complex on the surface of the bottom part of the primary cillium. Defects of PC-1 or PC-2, due to mutations in their respective PKD genes, lead to the deregulation of calcium homeostasis, mediated by the cAMP pathway. In one scenario, a complex of both polycystins could act as a mechanoreceptor signaling via Ca2+ influx through PC-2, a TRP channel, after fluid flow registration by PC-1 [7]. On the basis of recent results, the polycystin complex is associated with various cell signaling pathways (c-Myc, Wnt/β-catenin, hedgehog) [8,9,10]. Looking for new prognostic biomarkers as well as therapeutic targets and procedures is the constant ongoing approach [11,12,13,14,15].
Disease severity in ADPKD is directly related to the gene mutated and to the type of mutation found. Truncating variants of the PKD1 gene lead to ESRD at approximately 55.6 years of age; whereas nontruncating PKD1 mutations at 67.9 years of age. In the case of PKD2 variants, the average age of ESRD is approximately 79.7 years [16, 17]. However, the phenotypic spectrum is often significantly variable within a single family, and ranges from rare severe in utero-onset cases to patients with a preserved glomerular filtration rate into old age. This variability cannot be solely explained by the genetic heterogeneity caused by the PKD1 and PKD2 genotypes. Pronounced genic effects upon the clinical manifestation of ADPKD (early onset) have been observed in cases of contiguous deletion of PKD1 and TSC2 (OMIM *191092), in cases of mosaicism, and/or in cases of bilineal inheritance of a pathogenic PKD1 and PKD2 variant [18,19,20]. The latest clinical observations and animal studies have stressed the importance of hypomorphic alleles, which individually or in combination with other genes, can influence the severity of the ADPKD phenotype [21].
On the basis of current investigations, very mild PKD phenotypes, particularly in connection with significant polycystic liver disease (PLD), might be the result of a mutated GANAB gene (OMIM *104160; coding for glucosidase II subunit α), whose involvement is estimated at approximately 0.3% of total ADPKD [22].
Herein, we report a case of an ADPKD family, in which the youngest male member was referred to the department of nephrology in the age of twenty with an uremic syndrome and ESRD due to ADPKD, and in which bilineal inheritance of pathogenic PKD1 and PKD2 variants has been revealed based on genetic studies. We characterize the genic background in the proband and his family members, and discuss the genotype-phenotype correlations.
Case presentation
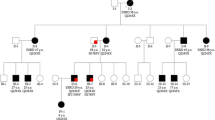
The proband presented soon after birth with enlarged kidneys filled with multiple cysts. A diagnosis of ADPKD was established based on the clinical picture and positive family history (ADPKD had been found in both father and older sister) (Fig. 1). At the age of 2 he experienced renal colic and acute renal failure, successfully treated conservatively. From the age of 6 he suffered from arterial hypertension, and at the age of 14 chronic renal insufficiency developed. Chronic kidney disease progressed to chronic renal failure at the age of 23. At that age he started hemodialysis treatment and was put on the waiting list for transplantation. After 1 year of hemodialysis treatment he underwent a successful cadaveric kidney transplantation with the graft’s function lasting over 15 years. After this period, he has returned to chronic dialysis treatment. Except for the renal symptoms related to ADPKD, the proband presented with local gigantism (macrodactyly) of the second toe on the left lower limb, which was significantly enlarged. Concurrently, the mental abilities of the proband and his younger sister (without ADPKD) are moderately below-average (they both attended a special school in childhood). Nevertheless, they have achieved writing and reading skills, and displayed sufficient mental and communicative capacities to integrate relatively smoothly into their milieu. Lowered intelligence was not observed in the case of the proband’s older sister, suffering from a mild form of ADPKD. The proband’s 80 year-old father suffers from moderate renal insufficiency (chronic kidney disease stage 3b, eGFR 33 ml/min/m2 according to CKD EPI formula) because of polycystic kidneys. He has cysts in his liver, as well. On echocardiography, significant pulmonary and tricuspidal insufficiency was described, with intermittent symptoms of right cardiac failure. The proband’s 52 year-old sister is being treated for polycystic kidneys and liver. Although she was repeatedly hospitalized for infections of renal cysts, her renal function is normal (eGFR 93.6 ml/min/m2).

Segregation of the disease in a family. The black-filled individuals, suffering from ADPKD, are marked with a blue arrow (the carriers of PKD2 mutation), and a red arrow (the carrier of PKD1 mutation). The dead woman with a dashed red arrow is the expected carrier of PKD1 mutation. The green arrow indicates the two members of the family with moderately lowered mental abilities. The proband is marked with a black arrow
The genomic DNA of all available family members was isolated from peripheral blood lymphocytes following standard procedures. At first, the mutational screening of the PKD2 gene was carried out within the running scientific project. Because of the mild clinical course of all affected living members (except for the proband), the presence of a PKD2 causative mutation in the family was highly expected. Mutation analysis was performed as previously described, using heteroduplex analysis and Sanger sequencing [23]. Indeed, a novel missense PKD2 mutation p.Arg420Gly was found in the proband, who was already dialyzed at that time (reference sequence NM_000297.3). The same mutation was confirmed in other available affected family members (the proband’s father, proband’s aunt, and her son), and later also in the proband’s older sister (see Fig. 1). The co-segregation of mutation with the disease in the family was proven and concurrently tested in 100 unrelated individuals, where no positive capture was observed. The substitution was tested using the verifying methods available to date, and defined as pathogenic.
Unfortunately, the finding of the PKD2 mutation couldn’t explain the serious ADPKD phenotype of the proband. For that reason, the seeking out of other affected family members was initiated. Finally, it was discovered that the proband’s mother had already died of renal failure at the age of 45 because of unrecognized polycystic kidney disease only established post mortem. Unfortunately, the DNA of the proband’s dead mother was not available for further examination; this also was the case for the DNA of the other living affected family members, because the proband’s mother had come from an orphanage. Nevertheless, concerning the early death of the proband’s mother, it was highly probable that she suffered from ADPKD type 1, and thus passed the pathogenic PKD1 sequence variant on to her son. Therefore, a mutational analysis of PKD1 of the proband was performed in order to identify the causative mutation of this gene. The experimental strategy was based on long-range PCR, followed by nested PCR and High Resolution Melting analysis (HRM). All nested PCR fragments showing aberrant melting curves on HRM were verified by Sanger sequencing in both directions. The entire procedure (including primers and PCR reaction conditions) was described in detail by Reiterová et al. [19]. As a result, the novel nonsense PKD1 mutation p.Gln2196* with a definitely pathogenic effect was identified (reference sequence NM_001009944.2). This mutation was subsequently published by Obeidova et al. [24]. All available family members (together with the proband’s older sister) were tested for the presence of this new PKD1 mutation; however, it was not found in any of them. The proband was thus the only living person in this family who carried the pathogenic nonsense PKD1 mutation. Besides, he also possessed the previously identified causative PKD2 mutation. Co-inheritance of both of these sequence variants of both PKD genes, and their common impact on ADPKD manifestations, are the presumptive explanation for such a complicated clinical course.
Interestingly, the same nonsense PKD1 mutation (p.Gln2196*) recently has been identified in another Czech family (in three women from three generations). The age of ESRD in the first and the second generation were 63 and 51, respectively. The third generation patient (32 years old) is being treated for hypertension. No additional PKD sequence variant has been found in this family up to the present. Some relationship between both of the families mentioned can not be ruled out.
The lower mental capabilities of the proband and his younger sister suggest a possible genetic load within the family. To solve this hypothesis, a Multiplex Ligation-dependent Probe Amplification (MLPA) analysis was carried out using a SALSA MLPA P245 Microdeletion Syndrome-1 kit, a SALSA MLPA P036 Subtelomeres Mix 1 kit, and a SALSA MLPA P070 Subtelomeres Mix 2B kit (all from MRC-Holland). The results of the MLPA were carried out by fragmentation analysis with the use of an ABI PRISM® 3130 Genetic Analyzer (Applied Biosystems). To obtain the final results, the raw data generated were normalized according to the manufacturer’s instructions. An in-house developed method was used for all calculations. The MLPA analysis was done with the DNA of our proband, and with the DNA of his older sister with normal intelligence (negative control). The DNA of the proband’s younger sister, with lower mental abilities (but without ADPKD) was unfortunately unavailable. The results of all three MLPA analyses were negative in both samples; no deletion or any other aberration was detected. As another step in our examination, the DNA of the proband was put through whole-genome screening by the method of Comparative Genomic Hybridization (array CGH) (Agilent, SurePrint G3 CGH ISCA v2 Microarray 8x60K) with the following specifications: CytoGenomics Edition 4.0.3.12, Agilent software; whole-genome screening with 240 kb resolution (48 kb in ISCA); Human genome GRCh37/hg19 database. As in the case of MLPA, no significant deletion/duplication referring to the corresponding phenotype was discovered. However, several large-scale studies have demonstrated that array CGH has a 10–20% detection rate of chromosomal abnormalities in children with mental retardation/developmental delay, with or without congenital anomalies [25]. Thus, a hereditary origin of the low mental abilities of the proband and his younger sister still can’t be excluded.
Discussion and conclusions
For the first time, within the Czech cohort of patients, we report on the bilineal inheritance of a PKD1 and PKD2 mutation within a family with ADPKD. The PKD1 and PKD2 genes, which are two basic genetic determinants of ADPKD, were localized in the early ‘90s of the previous century [4, 5]. Recently, the real genetic background of this disease has been found to be much more complicated. ADPKD shows significant inter- and intrafamilial phenotypic variability in the rate of disease progression and extra-renal manifestations, which can hardly be explained by the presence of single pathogenic mutation in the causal gene. In the vast majority of cases, mild phenotypes are related to the PKD2 gene. On the other hand, severe ADPKD manifestations, already frequently diagnosed in utero or at birth, have also been described [21, 26, 27]. These findings not only suggest the role of the PKD1 and PKD2 causative genes themselves, but also the involvement of heritable so-called modifier genes. Thus, the most severe PKD phenotypes are supposed to be a consequence of the concurrent impact of two co-inherited mutations; the first in the causative PKD1 gene, and the second in a modifier gene [20, 28]. Moreover, the second mutation of the modifier gene itself need not necessarily have the causative effect; a similar impact on the resulting deteriorated condition of an actual patient (family) may have the presence of either a hypomorphic allele (incompletely penetrant allele or allele, causing only partial loss of gene function) or an allele carrying polymorphisms or sequence variants of so far unknown significance (and acting both in cys or in trans) [19, 21, 26, 29, 30]. The common effect of two PKD1 hypomorphic alleles (in a homozygous or in a compound heterozygous state) on the development of ADPKD has also been described [31]. All of the main causative genes concerning polycystic kidney disease in general (PKD1, PKD2, PKHD1 as the main determinant of ARPKD or HNF-1β) in some circumstances can act as modifier genes and/or hypomorphic alleles [32]. Different combinations of causative and modifier genes thus probably ensure the enormous variability of ADPKD phenotypes. In reference to the PKD1 gene in general, the inherited modifiers and genetic background are supposed to account for an estimated 18 to 59% of the phenotypic variability [33].
In the case of the patient reported in this present publication, both mutations found in the PKD1 and PKD2 genes were described as highly likely to be pathogenic, and thus both with the highest probability have a causative effect [23, 24]. In the first place, the nonsense PKD1 mutation p.Gln2196* (probably inherited from the proband’s mother) is responsible for the unfavorable ADPKD type 1 phenotype, which is strongly aggravated by the presence of the second missense PKD2 mutation p.Arg420Gly. Despite the fact that we unfortunately don’t have a precise clinical characterization of the proband’s mother, as probably the only family representative simply having a PKD1 mutation alone, the disease manifestation in the proband is demonstrably much worse than the ordinary phenotype of ADPKD type 1 (ESRD at the age of 23 in the case of the proband, versus mid fifties for ADPKD type 1 in general). The finding of the second Czech family with a p.Gln2196* mutation of PKD1 (ESRD at the age of 63 and 51) is in full accordance with this assumption.
The first documentation of bilineal disease in ADPKD was published by Pei et al. in 2001, and completed by the same author 11 years later [20, 34]. There the authors described two ADPKD patients (members of a multigeneration family) with trans-heterozygous mutations in both the PKD1 and PKD2 genes. Molecular analysis of PKD2 revealed frameshift mutation p.Asn720fs; the presence of the second pathogenic mutation within PKD1 (p.Tyr528Cys) was proven later on, as itself causing a mild ADPKD phenotype. Also, in the case of these two patients with bilineal inheritance (ESRD at the age of 48 and 52), their ADPKD phenotype was more severe in comparison with other family members carrying only one type of PKD mutation. The clinical testing of two patients with trans-heterozygous PKD1 and PKD2 mutations showed severely reduced creatinine clearance when compared with patients who had only the PKD1 mutation, as well as more liver cysts than other affected family members, who had either PKD1 or PKD2 mutations alone. However, none of the two patients discussed had massive polycystic liver, resulting in either portal hypertension or hepatic failure. Also, none of the affected family members had any evidence of intracranial arterial aneurysms.
Another case of co-inheritance of the PKD1 and PKD2 mutation in trans was reported by Dedoussis et al. [28]. The proband of this study was a woman, diagnosed at the age of 39, suffering from hypertension and multiple cysts in both kidneys (without extrarenal cysts and without vascular complications and aneurysms). She reached ESRD at the age of 41, and at the age of 51 underwent successful kidney transplantation (from a cadaver donor). Again, ADPKD manifestation of this proband is thus demonstrably more serious than ADPKD caused by only a single mutation of PKD1. Detailed genetic analysis of the proband’s DNA revealed a novel homozygous missense mutation of PKD2 (p.Phe482Cys) and a de novo heterozygous PKD1 splice-site mutation (c.8017-2_-1delAG in IVS21). The pathogenicity of the novel PKD2 substitution was backed up with some experimental studies concerning protein (PC-2) expression, localization, and function.
The disease-modifying role of the non-causative missense variant was recently demonstrated by Ali et al. [30]. In that report, it was the PKD1 itself, which played the role of causative gene, and at the same time the role of modifying allele with a non-causative sequence variant acting in trans. In the four-generation’s family, the nonsense PKD1 mutation p.Gln2243* was determined, which co-segregated with the disease in the family, showing the typical ADPKD type 1 phenotype. Two members of that family (brothers), except for the nonsense mutation mentioned above, carried another PKD1 sequence variant (p.His1769Tyr), evaluated as a variant of undetermined significance using different computer prediction tools. Both brothers inherited this new sequence variant from their healthy father. The first proband carrying both the causative and the undetermined PKD1 sequence variant had already reached ESRD at the age of 29. His brother, with the same PKD1 genotype, showed a similar extreme enlargement of the kidneys, although his renal function was in the normal range. The undetermined sequence variant itself had only a very mild effect on disease manifestation, as was indicated by the case of the healthy father of the two affected brothers. The father carried only the undetermined variant without any PKD1 causative mutation. Although he was considered to be healthy, had normal kidney volume, normal kidney function, and no family history of ADPKD, an ultrasound examination revealed seven renal cysts at the age of 58.
A similar case of a bilineal ADPKD family with two PKD1 mutations/sequence variants in trans (in-frame deletion and substitution) has been recently reported by Hwang et al. [35].
Not only the genes directly determining the polycystic kidney disease (ADPKD or ARPKD) can act as modifiers in the development of ADPKD. The modifying effect and related aggravated disease phenotype also has recently been described in connection with the TTC21B gene determining other kinds of ciliopathies (viz., nephronophthisis and focal segmental glomerulosclerosis) [36].
More genetic variants elsewhere in the genome, modifying the progression of ADPKD, could be further revealed by current methods of molecular genetics, such as Next-Generation Sequencing (NGS) or Genome-Wide Association Study (GWAS) [29, 37,38,39].
In conclusion, here we described the first ADPKD family in the Czech Republic, with an atypical clinical course of one family member, caused by bilineal inheritance of pathogenic PKD1 and PKD2 variants. The presented report illustrates the fact that not only the single gene (PKD1 or PKD2) is responsible for the manifestation of ADPKD. Except for the causative gene, the final ADPKD phenotype is also modulated by renal or alternatively non-renal modifier genes, hypomorphic alleles, alleles carrying different (especially SNP) polymorphisms, and by the genetic background, in general. Co-inheritance of two or more genes/alleles in connection with ADPKD appears not to be as rare as previously considered. On the contrary, this is probably one of the main mechanisms explaining the existing vast inter- and intrafamilial variability in the ADPKD phenotype.
Abbreviations
- ADPKD:
-
Autosomal dominant polycystic kidney disease
- ARPKD:
-
Autosomal recessive polycystic kidney disease
- CGH:
-
Comparative genomic hybridization
- ESRD:
-
End stage renal disease
- HRM:
-
High resolution melting
- MLPA:
-
Multiplex ligation-dependent probe amplification
- PC-1:
-
Polycystin 1
- PC-2:
-
Polycystin 2
- PKD:
-
Polycystic kidney disease
- PLD:
-
Polycystic liver disease
References
Cordido A, Besada-Cerecedo L, García-González MA. The genetic and cellular basis of autosomal dominant polycystic kidney disease-a primer for clinicians. Front Pediatr. 2017;5:279.
Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329(5):332–42.
Chapman AB, Devuyst O, Eckardt KU, Gansevoort RT, Harris T, Horie S, et al. Autosomal dominant polycystic kidney disease (ADPKD): executive summary from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. 2015;88(1):17–27.
European Polycystic Kidney Disease Consortium. The polycystic kidney disease I gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;77:881–94.
Kimberling WJ, Kumar S, Gabow PA, Kenyon JB, Connolly CJ, Somlo S. Autosomal dominant polycystic kidney disease: localization of the second gene to chromosome 4q13-q23. Genomics. 1993;18:467–72.
Tan YC, Blumenfeld J, Rennert H. Autosomal dominant polycystic kidney disease: genetics, mutations and microRNAs. Biochim Biophys Acta. 2011;1812(10):1202–12.
Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–37.
Trudel M. C-Myc Signalling in the genetic mechanism of polycystic kidney disease. In: Li X, editor. Polycystic kidney disease. Brisbane: Codon Publications; 2015. Chapter 10.
Kim S, Nie H, Nesin V, Tran U, Outeda P, Bai CX, et al. The polycystin complex mediates Wnt/ca(2+) signalling. Nat Cell Biol. 2016;18(7):752–64.
Habbig S, Liebau MC. Ciliopathies - from rare inherited cystic kidney diseases to basic cellular function. Mol Cell Pediatr. 2015;2(1):8.
Riella C, Czarnecki PG, Steinman TI. Therapeutic advances in the treatment of polycystic kidney disease. Nephron Clin Pract. 2014;128(3–4):297–302.
Saigusa T, Bell PD. Molecular pathways and therapies in autosomal-dominant polycystic kidney disease. Physiology (Bethesda). 2015;30(3):195–207.
de Stephanis L, Bonon A, Varani K, Lanza G, Gafà R, Pinton P, et al. Double inhibition of cAMP and mTOR signalling may potentiate the reduction of cell growth in ADPKD cells. Clin Exp Nephrol. 2017;21(2):203–11.
Streets AJ, Magayr TA, Huang L, Vergoz L, Rossetti S, Simms RJ, et al. Parallel microarray profiling identifies ErbB4 as a determinant of cyst growth in ADPKD and a prognostic biomarker for disease progression. Am J Physiol Renal Physiol. 2017;312(4):F577–88.
Wong ATY, Mannix C, Grantham JJ, Allman-Farinelli M, Badve SV, Boudville N, et al. Randomised controlled trial to determine the efficacy and safety of prescribed water intake to prevent kidney failure due to autosomal dominant polycystic kidney disease (PREVENT-ADPKD). BMJ Open. 2018;8(1):e018794.
Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24(6):1006–13.
Cornec-Le Gall E, Audrézet MP, Renaudineau E, Hourmant M, Charasse C, Michez E, et al. PKD2-related autosomal dominant polycystic kidney disease: prevalence, clinical presentation, mutation Spectrum, and prognosis. Am J Kidney Dis. 2017;70(4):476–85.
Consugar MB, Wong WC, Lundquist PA, Rossetti S, Kubly VJ, Walker DL, et al. Characterization of large rearrangements in autosomal dominant polycystic kidney dinase and the PKD1/TSC2 contiguous gene syndrome. Kidney Int. 2008;74:1468–79.
Reiterová J, Štekrová J, Merta M, Kotlas J, Elišáková V, Lněnička P, et al. Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol. 2013;14:59.
Pei Y, Paterson AD, Wang KR, He N, Hefferton D, Watnick T, et al. Bilineal disease and trans heterozygotes in autosomal dominant polycystic kidney disease. Am J Hum Genet. 2001;68:355–63.
Vujic M, Heyer CM, Ars E, Hopp K, Markoff A, Orndal C, et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. JAm Soc Nephrol. 2010;21:1097–102.
Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, et al. Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet. 2016;98(6):1193–207.
Stekrová J, Reiterová J, Merta M, Damborsky J, Zidovská J, Kebrdlová V, et al. PKD2 mutations in a Czech population with autosomal dominant polycystic kidney disease. Nephrol Dial Transplant. 2004;19(5):1116–22.
Obeidova L, Elisakova V, Stekrova J, Reiterova J, Merta M, Tesar V, et al. Novel mutations of PKD genes in the Czech population with autosomal dominant polycystic kidney disease. BMC Med Genet. 2014;15:41.
Moorchung N. Molecular techniques in preimplantation genetic diagnosis. In: Talwar CP, editor. Manual of Cytogenetics in Reproductive Biology. Jaypee Brothers Medical Publishers; 2014. p.73-86.
Bergmann C, von Bothmer J, Ortiz Brüchle N, Venghaus A, Frank V, Fehrenbach H, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol. 2011;22:2047–56.
Bergmann C. ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol. 2015;30(1):15–30.
Dedoussis GV, Luo Y, Starremans P, Rossetti S, Ramos AJ, Cantiello HF, et al. Co-inheritance of a PKD1 mutation and homozygous PKD2 variant: a potential modifier in autosomal dominant polycystic kidney disease. Eur J Clin Investig. 2008;38(3):180–90.
Harris PC, Rossetti S. Determinants of renal disease variability in ADPKD. Adv Chronic Kidney Dis. 2010;17(2):131–9.
Ali H, Hussain N, Naim M, Zayed M, Al-Mulla F, Kehinde EO, et al. A novel PKD1 variant demonstrates a disease-modifying role in trans with a truncating PKD1 mutation in patients with autosomal dominant polycystic kidney disease. BMC Nephrol. 2015;16:26.
Rossetti S, Kubly VJ, Consugar MB, Hopp K, Roy S, Horsley SW, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75(8):848–55.
Audrézet MP, Corbiere C, Lebbah S, Morinière V, Broux F, Louillet F, et al. Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27(3):722–9.
Fain PR, McFann KK, Taylor MR, Tison M, Johnson AM, Reed B, et al. Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney Int. 2005;67(4):1256–67.
Pei Y, Lan Z, Wang K, Garcia-Gonzalez M, He N, Dicks E, et al. A missense mutation in PKD1 attenuates the severity of renal disease. Kidney Int. 2012;81(4):412–7.
Hwang YH, Conklin J, Chan W, Roslin NM, Liu J, He N, et al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27(6):1861–8.
Bullich G, Vargas I, Trujillano D, Mendizábal S, Piñero-Fernández JA, Fraga G, et al. Contribution of the TTC21B gene to glomerular and cystic kidney diseases. Nephrol Dial Transplant. 2017;32(1):151–6.
Eisenberger T, Decker C, Hiersche M, Hamann RC, Decker E, Neuber S, et al. An efficient and comprehensive strategy for genetic diagnostics of polycystic kidney disease. PLoS One. 2015;10(2):e0116680.
Kinoshita M, Higashihara E, Kawano H, Higashiyama R, Koga D, Fukui T, et al. Technical evaluation: identification of pathogenic mutations in PKD1 and PKD2 in patients with autosomal dominant polycystic kidney disease by next-generation sequencing and use of a comprehensive new classification system. PLoS One. 2016;11(11):e0166288.
Liu M, Shi S, Senthilnathan S, Yu J, Wu E, Bergmann C, et al. Genetic variation of DKK3 may modify renal disease severity in ADPKD. J Am Soc Nephrol. 2010;21(9):1510–20.
Acknowledgements
We are grateful to all of the patients and their family. We thank Dr. Jana Lastuvkova, Dr. Marketa Urbanova PhD., Martina Reznickova, and Monika Skodova for their valuable cooperation.
Funding
This work was supported by grant projects IGA MZ CR: NE 7633, IGA MZ CR: NR 9427, Charles University: PROGRES Q25/LF1 and GAUK 1015.
Availability of data and materials
All datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
VE drafted the manuscript and carried out the molecular genetic studies. MM drafted the manuscript, collected data, and evaluated the clinical diagnosis. JR carried out the molecular genetic studies, collected data, and evaluated the clinical diagnosis. AB and JK consulted patients as clinical geneticists, and helped with examinations of relatives. KH carried out the MLPA analysis. LO and VT participated in the conception and design of the study. JS designed the study, and performed the molecular genetic analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of General University Hospital in Prague. All examined patients provided written informed consent for genetic testing.
Consent for publication
Written informed consent for participation in the anonymous research as well as for publication of its results (together with any related accompanying images) was obtained from all participating family members.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Elisakova, V., Merta, M., Reiterova, J. et al. Bilineal inheritance of pathogenic PKD1 and PKD2 variants in a Czech family with autosomal dominant polycystic kidney disease – a case report. BMC Nephrol 19, 163 (2018). https://doi.org/10.1186/s12882-018-0978-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-018-0978-2