
Abstract
Background
Thrombotic microangiopathy (TMA) is a serious, sometimes life-threatening disorder marked by the presence of endothelial injury and microvascular thrombi. Drug-induced thrombotic microangiopathy (DI-TMA) is one specific TMA syndrome that occurs following drug exposure via drug-dependent antibodies or direct tissue toxicity. Common examples include calcineurin inhibitors Tacrolimus and Cyclosporine and antineoplastics Gemcitabine and Mitomycin. Valproic acid has not been implicated in DI-TMA. We present the first case of a patient meeting clinical criteria for DI-TMA following admission for valproic acid toxicity.
Case presentation
An adolescent male with difficult to control epilepsy was admitted for impaired hepatic function while on valproic acid therapy. On the third hospital day, he developed severe metabolic lactic acidosis and multiorgan failure, prompting transfer to the pediatric intensive care unit. Progressive anemia and thrombocytopenia instigated an evaluation for thrombotic microangiopathy, where confirmed by concomitant hemolysis, elevated lactate dehydrogenase (LDH), low haptoglobin, and concurrent oliguric acute kidney injury. Thrombotic thrombocytopenic purpura was less likely with adequate ADAMTS13. Discontinuing valproic acid reversed the anemia, thrombocytopenia, and normalized the LDH and haptoglobin, supporting a drug-induced cause for the TMA.
Conclusion
To the best of our knowledge, this is the first report of drug-induced TMA from valproic acid toxicity.
Similar content being viewed by others


Background
The diagnosis of drug-induced thrombotic microangiopathy (DI-TMA) is often complex and difficult. TMAs are a group of heterogeneous disorders defined not only by the presence of microvascular thrombi, thrombocytopenia and hemolysis involving predominantly the kidney but all organ systems [1, 2]. Considered a prothrombotic condition secondary to an underlying disturbance of coagulation and complement systems, etiologies include: thrombotic thrombocytopenic purpura (TTP), a systemic disorder of microvascular thrombosis of ADAMTS13 deficiency, hemolytic uremic syndrome (HUS), with and without complement mutations, characterized by nonimmune hemolytic anemia, thrombocytopenia, and renal failure, and drug induced TMA (DI-TMA), through either a direct toxic effect or duration dependent toxicity. [3]
Valproic acid (VPA), an anticonvulsive agent for epileptic syndromes, has adverse consequences associated with its use; most mild and transient but others serious, affecting embryonic development and still others, uncommonly, causing significant disorders of hepatic and pancreatic dysfunction, Fanconi syndrome and weight gain [4]. Hematological disorders are seen in children treated with VPA, including thrombocytopenia, platelet dysfunction, Von Willebrand disease, Factor XIII deficiency, hypofibrinogenemia and vitamin K-dependent factor deficiency. A dual etiology is responsible: direct toxicity of VPA on bone marrow and VPA’s inclusion with and modification of the platelet membrane [4].
Acute TMA in conjunction with VPA toxicity has, to our knowledge, not been reported. We propose that VPA’s ability to modify cellular membranes may explain the TMA and acute kidney injury seen in our patient.
Case presentation
A 16 year old male with epilepsy and variable response to anti-convulsants presented to our hospital. Previously controlled with a combination of valproic acid (VPA) and levetiracetam for many years, his seizures recurred despite high therapeutic levels, prompting dose modifications. At a routine clinic appointment, he complained of severe emesis, prompting admission. There his VPA level was low, 46 μg/ml (normal range 50–100 μg/ml). He was bolused with VPA (15 mg/kg), achieving a toxic VPA level of 137 μg/ml, remaining elevated (130 μg/ml) for 16 h. The typical half-life for VPA is 9–16 h in adolescents. His ALT (234 unit/L) and AST (63 unit/L) were mildly elevated so intravenous (IV) L-carnitine was initiated for VPA-induced hepatotoxicity.
Subsequently an abrupt deterioration manifested, a severe anion gap metabolic acidosis (pH 7.05, pCO2 24 mmHg, serum bicarbonate 12 mEq/L, serum lactic acid 16.9 mMol/L), and acute kidney injury (creatinine increased from 1.2 mg/dL on admission to 2.4 mg/dL). Now in the pediatric intensive care unit, despite adequate volume resuscitation and correction of pH, his creatinine rose to 2.6 mg/dL.
Laboratory findings concomitantly consistent with a TMA: acute kidney injury, anemia (hemoglobin 9.1 to 8.1 g/dL), thrombocytopenia (platelets 25,000 K/CMM), an elevated LDH (350 unit/L) (normal range 98–192 unit/L) and suppressed haptoglobin (<8 mg/dL) with reticulocyte count of 6.5%, reflecting adequate bone marrow response, all supported a diagnosis of acute hemolytic anemia. Peripheral blood smear showed mild macrocytic anemia, mild anisopoikilocytosis with target cells and rare schistocytes, platelets were markedly decreased. ADAMTS 13 activity reported 59% enzyme function (normal >67%). Complement levels, low C3 55 mg/dL (normal 88–201) and normal C4 30 mg/dL, suggested alternative pathway activation.
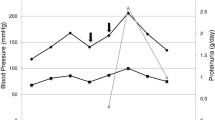
Following discontinuation of VPA, levels normalized in five days. Concurrently, his serum creatinine, serum bicarbonate, hemoglobin and haptoglobin improved to normal ranges (Fig. 1). Complement evaluation was entertained, but ultimately not pursued when C3 normalized to 159 mg/dL after medication discontinuation. His anti-epileptic regimen was changed to Vimpat (lincosamide) and Keppra (levetiracetam), and he remained free of TMA.

Laboratory values during hospitalization
Discussion
Valproic acid is an effective anticonvulsive agent with several adverse consequences. Thrombocytopenia is the most common hematologic adverse effect, with incidence varying from 5% to 60% [5]. In 2007, Beydoun and Nasreddine illustrated the association between VPA toxicity and thrombocytopenia. The probability of developing thrombocytopenia increases when trough VPA levels are above 130 μg/ml [6]. While our patient’s prolonged VPA level elevation above 130 μg/ml are consistent with these studies, the microangiopathic hemolytic anemia, anuria, and acute kidney injury reflect a more severe hematologic etiology, consistent with thrombotic microangiopathy (TMA) syndrome.
TMA syndromes are extraordinarily diverse, divided into hereditary and acquired etiologies. Universally, TMA syndromes are incited by microvascular endothelial injury leading to arteriolar and capillary thrombosis and subsequent organ injury. DI-TMA occurs via 2 mechanisms, either non-dose related immunologic reactions or toxic-dose related reactions. While toxic-dose mediated TMA is gradual in onset, renal failure can be seen over weeks to months, immune-reaction TMA is more sudden [7].
Drug-mediated immune reactions leading to TMA were first recognized in 1980 after documented exposures to quinine [7]. Quinine antibodies included immunoglobulin G (IgG) or IgM, reactive with either glycoprotein Ib/IX or IIb/IIIa or both [8, 9]. Antibodies against granulocytes, lymphocytes, and endothelial cells have been described [9].
VPA, a branched-chain carboxylic acid, is structurally and chemically similar to fatty acid constituents of cell membranes. Sandler et al. proposed this similarity would lead to production of immunoglobulin M (IgM) antibodies against circulating thrombocytes with decreasing antibody titers after recovery [5]. For many years, Sandler’s proposal remained hypothetical. Barr et al. affirmed Sandler’s proposal when he documented increased levels of IgG platelet antibodies in half of patients receiving VPA, reflecting increased platelet destruction [10].
VPA is also shown to affect the mean corpuscular volume (MCV) of erythrocytes. Konig et al. studied 3 anticonvulsant therapies (valproic acid, carbamazepine, or phenobarbital) effect on erythrocyte cell membranes. VPA was the only anti-epileptic drug (AED) to alter the fatty acid composition of erythrocytes [11]. Affecting membrane fluidity and altering receptor proteins expressed on both the inner and outer surfaces of the erythrocyte cell membrane provides the nidus for immunoglobulin directed destruction of erythrocytes.
We performed a literature review searching for prior documentation of TMA-associated with VPA therapy. Results revealed 4 papers, three reporting isolated thrombocytopenia cases with VPA toxicity as previously mentioned. The fourth highlighted TTP and anemia in a breast fed healthy male infant whose mother was treated with VPA, unique for two reasons. First, a simultaneous decrease in both platelets and hemoglobin, not typical of either thrombocytopenia from VPA or idiopathic thrombocytopenic purpura (ITP). Secondly, no concentration-dependent toxic effect was seen since the infant’s serum VPA concentration remained low during the entire clinical course. Similar to our patient, thrombocytopenia and anemia resolved with removal of the drug, in this case cessation of breastfeeding. [12] After thorough medication review, an undetermined etiology makes VPA or its metabolites a possible culprit.
First recognized in 1979, there is increasing awareness of pancreatitis with disseminated intravascular coagulation (DIC) [13]. By July 2010, the FDA introduced new black box warnings for VPA, pancreatitis, however, the diagnosis of pancreatitis requires strict criteria. Explicitly, amylase and/or lipase elevation should be greater than 3 times the upper limit of normal, imaging studies should show evidence of inflammation, and, if performed, pancreatic damage should be visualized during surgery [14]. While still possible, our patient’s lab values and absent inflammation on imaging, make pancreatitis unlikely. Using the International Society for Thrombosis and Hemostasis DIC scoring system, our patient never met criteria for overt DIC [15].
Presenting with nausea/vomiting in up to 50% of patients, VPA hepatotoxicity occurs due to disruption of mitochondrial processes, proposed as drug-induced carnitine deficiency. As carnitine facilitates fatty acyl group transport into and out of the mitochondria, when acyl groups accumulate (as in drug intoxications), carnitine assists in transporting these acyl groups out of the mitochondria and eventually into the urine, inducing a secondary carnitine deficiency [16].
While rare and often fatal, timely initiation of intravenous L-carnitine treatment can often be life-saving. The patient’s neurologist began IV L-carnitine (50 mg/kg loading dose followed by 50 mg/kg over the next 24 h and then 100 mg/kg/day in divided doses for 7 days) within five days of admission, a favorable factor for improving hepatic function and VPA metabolism. Carnitine deficiency explains the impaired beta-oxidation of VPA allowing for immune-mediated TMA.
The presence of acute liver injury should not detract from suspicion for TMA. A subset of patients with endothelial damage have TAMOF, thrombocytopenia-associated multi-organ failure. This entity is increasingly identified in critically ill pediatric patients with signs consistent with TTP/HUS, but without significant hemolysis as seen by rare schistocytes, normal haptoglobin, and mildly suppressed ADAMTS13 activity. TAMOF exists within a spectrum of TMA syndromes, commonly defined by systemic endothelial cell destruction. Resolution of multiple organ dysfunction requires elimination of the source of inflammation [17, 18]. Swift recognition and VPA discontinuation likely saved this patient from escalation to plasma exchange and/or hemodialysis.
Further supporting our DI-TMA hypothesis, the Naranjo scale characterizes the likelihood of a true adverse drug reaction (ADR) as doubtful, possible, probable, and definite [19]. A score of 7 makes VPA a probable culprit, limited by lack of placebo and no prior conclusive reports. Secondary TMA from VPA toxicity associated with multiple organ failure is the consensus diagnosis. Our case features the first reported case of DI-TMA with VPA toxicity. We suggest increasing awareness of VPA as a TMA culprit will assist in identifying future cases.
Conclusion
DI-TMA may be a serious complication, previously unreported, of VPA therapy and therefore important to recognize. A prerequisite of successful treatment is the correct diagnosis which in this case was defined by exclusion. With ongoing TMA and AKI, despite drug cessation, consideration for a renal biopsy and anti-complement therapy should also be given. The possibility of adverse hematologic events secondary to VPA therapy should also be kept in mind.
Abbreviations
- DI-TMA:
-
Drug induced thrombotic microangiopathy
- HAP:
-
haptoglobin.
- LDH:
-
Lactate Dehydrogenase
- TMA:
-
thrombotic microangiopathy
- VPA:
-
valproic acid
References
Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Drug-induced thrombotic microangiopathy: a systematic review of published reports. Blood 2015 Jan 22;125(4):616–618. doi: 10.1182/blood-2014-11-611335. Epub 2014 Nov 20.
Reese JA, Bougie DW, Curtis BR, Terrell DR, Vesely SK, Aster RH, George JN. Drug-induced thrombotic microangiopathy: experience of the Oklahoma registry and the BloodCenter of Wisconsin. Am J Hematol 2015 May;90(5):406–410. Epub 2015 Feb 25.
Nadasdy T. Thrombotic microangiopathy in renal allografts: the diagnostic challenge. Curr Opin Organ Transplant. 2014, Jun;19(3):283–92.
Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. J Biomed Biotechnol, 2010. Article ID 479364, 18 pages. Epub 2010 Jul 29.
Gerstner T, Teich M, Bell N, Longin E, Dempfle CE, Brand J, König S. Valproate-associated coagulopathies are frequent and variable in children. Epilepsia. 2006 Jul;47(7):1136–43.
Nasreddine W, Beydoun A. Valproate-induced thrombocytopenia: a prospective monotherapy study. Epilepsia 2008 Mar;49(3):438–445. Epub 2007 Nov 21.
George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014 Nov 6;371(19):1847–8.
Stroncek DF, Vercellotti GM, Hammerschmidt DE, Christie DJ, Shankar RA, Jacob HS. Characterization of multiple quinine-dependent antibodies in a patient with episodice hemolytic uremic syndrome and immune agranulocytosis. Blood July 1992; 80:241–248.
Bougie DW, Wilker PR, Aster RH. Patients with quinine-induced immune thrombocytopenia have both “drug-dependent” and “drug-specific” antibodies. Blood April 2006; 108:922–927.
Barr RD, Copeland SA, Stockwell ML, Morris N, Kelton JC. Valproic acid and immune thrombocytopenia. Arch Dis Child. 1982 Sep;57(9):681–4.
König SA, Knolle J, Friedewald S, Koelfen W, Longin E, Lenz T, Hannak D. Effects of valproic acid, carbamazepine, and phenobarbitone on the fatty acid composition of erythrocyte membranes in children. Epilepsia. 2003 May;44(5):708–11.
Stahl MM, Neiderud J, Vinge E. Thrombocytopenic purpura and anemia in a breast-fed infant whose mother was treated with valproic acid. J Pediatr. 1997 Jun;130(6):1001–3.
Asconape JJ, Penry JK, Dreifuss FE, Riela A, Mirza W. Valproate-associated pancreatitis. Epilepsia. 1993;34:177–83.
Gerstner T, Büsing D, Bell N, Longin E, Kasper JM, Klostermann W, Hebing B, Hanefeld F, Eckel U, Hoffmann R, Bettendorf U, Weidner B, Wiemer-Kruel A, Brockmann K, Neumann FW, Sandrieser T, Wolff M, König S. Valproic acid-induced pancreatitis: 16 new cases and a review of the literature. J Gastroenterol 2007 Jan;42(1):39–48. Epub 2007 Feb 16.
Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br J Haematol. 2009 Apr;145(1):24–33.
Bohan TP, Helton E, McDonald I, Konig S, Gazitt S, Sugimoto T, Scheffner D, Cusmano L, Li S, Koch G. Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurol. 2001;56:1405–9.
Nguyen TC, Carcillo JA. Bench to bedside review: thrombocytopenia-associated multiple organ failure- a newly appreciated syndrome in the critically ill. Crit Care. 2006;10:235.
Carcillo JA, Podd B, Aneja R, Weiss SL, Hall MW, Cornell TT, Shanley TP, Doughty LA, Nguyen TC. Pathophysiology of pediatric multiple organ dysfunction syndrome. Pediatr Crit Care Med. 2017;18:3.
Naranjo CA, Busto U, Sellers EM, Sandor P, Ruiz I, Roberts EA, Janecek E, Domecq C, Greenblatt DJ. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239–45.
Acknowledgments
This study was not funded by any third party.
Funding
Not Applicable.
Availability of data and materials
Not Applicable.
Author information
Authors and Affiliations
Contributions
SH performed the literature review, wrote the manuscript, and was a treating physician for the patient. RS assisted drafting the manuscript and was a treating physician for the patient. CE assisted with drafting the manuscript and was a treating physician for the patient. TB assisted with the literature review and was a treating physician for the patient. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained by the patient and family for publication in this Case report. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
Dr. Swinford would like to declare she is a speaker for Alexion Pharmaceuticals, Inc. The remaining authors declare no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hebert, S.A., Bohan, T.P., Erikson, C.L. et al. Thrombotic microangiopathy associated with Valproic acid toxicity. BMC Nephrol 18, 262 (2017). https://doi.org/10.1186/s12882-017-0677-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-017-0677-4