
Abstract
Background
Damaging variants in TRIO have been associated with moderate to severe neurodevelopmental disorders in humans. While recent work has delineated the positional effect of missense variation on the resulting phenotype, the clinical spectrum associated with loss-of-function variation has yet to be fully defined.
Case presentation
We report on two probands with novel loss-of-function variants in TRIO. Patient 1 presents with a severe neurodevelopmental disorder and macrocephaly. The TRIO variant is inherited from his affected mother. Patient 2 presents with moderate developmental delays, microcephaly, and cutis aplasia with a frameshift variant of unknown inheritance.
Conclusions
We describe two patients with neurodevelopmental disorder, macro/microcephaly, and cutis aplasia in one patient. Both patients have loss-of-function variants, helping to further characterize how these types of variants affect the phenotypic spectrum associated with TRIO. We also present the third reported case of autosomal dominant inheritance of a damaging variant in TRIO.
Similar content being viewed by others

Background
The human gene TRIO (triple functional domain) encodes a guanine nucleotide exchange factor (GEF) that facilitates the activation of Rho GTPases such as RAC1 [1,2,3]. Rho GTPases in turn regulate actin cytoskeleton dynamics and thus play an important role in many neurodevelopmental processes such as migration of neural progenitors and neurons during neurogenesis, synapse formation, and axon outgrowth [4,5,6,7]. The TRIO protein contains several catalytic domains including two GEF domains. The GEF1 domain activates RAC1 and RHOG while the GEF2 domain activates RHOA [8,9,10,11]. The protein also contains a c-terminal serine/threonine kinase domain [3]. TRIO is a large gene with 57 exons and multiple tissue-specific isoforms, including several different isoforms expressed in the mammalian brain [12,13,14,15]. Studies have shown that expression of TRIO peaks in the brain in the late prenatal period [16] supporting a role for this gene in neurodevelopmental processes. The importance of this gene in mammalian neurodevelopment has been further highlighted by studies in mice which have shown complete knockout of the gene to be embryonic lethal, while nervous system-specific knockout mice show abnormal brain morphology, reduced brain size, and altered learning ability [17,18,19,20,21]. Additionally, it has been shown in vitro that TRIO is important in regulating synaptic function through regulation of AMPAr endocytosis at CA1 excitatory synapses [22].
Several patients have been identified with rare or novel variants in TRIO from large cohort studies examining whole exome sequencing data from patients with generalized phenotypes such as intellectual disability (ID), neurodevelopmental disorders, schizophrenia, childhood speech apraxia, autism spectrum disorder (ASD), microcephaly, and epilepsy with intellectual disability [16, 22,23,24,25,26,27,28,29,30,31,32,33,34,35]. Recently, however, several TRIO-specific cohorts with functional studies have been described that have helped elucidate the role of pathogenic variation in this gene in human disease [16, 22, 36,37,38]. Consistently reported phenotypes described in association with variants in this gene include global developmental delay, speech disorder, ID and learning disabilities, and macro- or microcephaly. Additional phenotypes include behavioral issues, dysmorphic facies, skeletal/hand anomalies, feeding difficulties, insensitivity to pain, seizures, tremor, ataxia, abnormal brain MRI, hypotonia, dental anomalies, congenital heart defects, urinary incontinence, and ocular defects.
Interestingly, it has become clear that there is a strong phenotype/genotype correlation specific to missense variants in this gene. Several missense variants that cluster in the spectrin repeat domain have been functionally shown to lead to increased RAC1 activation and enhanced neurite outgrowth and lamellipodia formation [36] (Fig. 1). Strikingly, all of the patients with missense variants in this region demonstrated moderate to severe ID and macrocephaly. Of note, Pengelly et al. also reported a patient with a p.Asn1080Ile missense variant who displayed severe ID but normal head circumference [37]. Their assessment of this variant in vitro did not identify an effect on the activation of RAC1, though this discrepancy with previous reports may be due to experimental design. Overall, it is hypothesized that missense variants in this domain cause hyperactivation of RAC1 leading to the observed macrocephaly phenotype. This may be due to variants affecting protein folding and thus interfering with the intermolecular inhibition of the GEF1 domain by the spectrin repeats [36, 39]. Conversely, missense variants in the RAC1 activating GEF1 domain have been shown functionally to lead to reduced overall RAC1 binding and activation, impaired formation of neurites and lamellipodia, and a reduction in AMPAR-eEPSC amplitude [16, 36,37,38] (Fig. 1). The phenotypes associated with these variants include mild/moderate ID and microcephaly. In contrast to variants in the spectrin domains, it is hypothesized that variants in the GEF1 domain lead to loss of normal RAC1 activation and thus the observed microcephaly. The reason for the severity of ID appears to be greater in association with spectrin variants is unclear. There has also been one report of a hypermorphic variant in the GEF2 domain in which functional studies showed increased RhoA activity [16] (Fig. 1). It is hypothesized that this domain is auto-inhibited by the PH2 domain, and variants that disrupt this inhibition lead to hyperactivity. The phenotype associated with this variant is not well defined other than schizophrenia.

Variants associated with neurodevelopment disorder in TRIO. Schematic of protein domains of human TRIO (NM_NM_007118.3). Variants discussed in this report are in black. Variants associated with microcephaly are in blue; microcephaly-associated frameshift/nonsense variants span the gene, while microcephaly-associated missense variants cluster in the GEF1 domain. Macrocephaly-associated missense variants cluster in the Spectrin repeats domain. With the exception of current patient 1 described here, all frameshift/nonsense variants have to date been associated with microcephaly
While clear associations have been shown between the location of missense variants and the resulting disease phenotype, the characterization of framsehift and nonsense variants is less well established. The majority of reported patients with loss-of-function (LOF) variants with occipitofrontal circumference (OFC) data available do display microcephaly. However, the severity of ID and other associated phenotypes does not appear to be consistent. In vitro modeling of truncating variants in the GEF1 domain has shown reduced RAC1 binding and activation [36,37,38], while modeling of truncating variants in the downstream GEF2 domain show show no effect on RAC1 activation. An open question remains as to whether or not these frameshift/nonsense variants are actually producing a truncated protein in vivo or if the transcripts are undergoing nonsense-mediated decay (NMD) resulting in haploinsufficiency. The presence of multiple alternate transcripts could also result in a variant-specific effect on NMD based on which transcripts are affected. Of the frameshift/nonsense described in TRIO cohorts so far, all would be predicted to undergo NMD based on an in silico prediction tool [40]. However none of the studies mentioned above have actually tested patient samples for absence or presence of a truncated protein product.
Here we present two patients presenting to the Department of Clinical Genomics at the Mayo Clinic with moderate/severe developmental delay, facial dysmorphisms, macro- and microcephaly, and a novel phenotype of cutis aplasia in one patient. Both patients were found to have presumed LOF variants in TRIO (Fig. 1). Additionally, one of the two probands harbors a variant inherited from an affected mother, making this the third documented cases of autosomal dominant (AD) familial inheritance of disease associated with this gene. The data from these patients help expand the known phenotypes associated with pathogenic TRIO variants, provides further evidence of AD inheritance, and helps elucidate the disease spectrum associated specifically with presumed LOF variants.
Case presentation
Clinical description
Patient 1 is a 14-year-old Caucasian boy who was born to a non-consanguineous union. He presented for genetic evaluation of autism spectrum disorder (ASD). He was born at term with appropriate birth weight, after an uncomplicated pregnancy and delivery. He had isolated delay in attaining socio-adaptive and language milestones and was noted to have repetitive behaviors. He remains nonverbal and also displays motor stereotypies such as hand flapping. Significant sleep disturbances were noticed since infancy. He was diagnosed with autism spectrum disorder at four years of age. He had febrile seizures during early childhood. He is currently in middle school with Individualized Education Program and communicates to some extent with augmented communication device. Significant behavioral anger outbursts occurred when he transitioned from elementary school to middle school which responded to symptomatic medications. There were no self-injurious behaviors or tactile sensitivities. His family history was significant for his mother with developmental delay and dyslexia as well as inflammatory bowel disease, one maternal uncle with significant developmental disability attributed to traumatic brain injury suffered at age 3, a paternal uncle with schizophrenia and a maternal 2nd cousin with autism spectrum disorder (Fig. 2b).

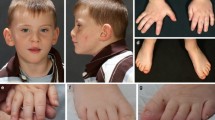
Characteristics of patients described with novel loss-of-function variants in TRIO. a Photograph of patient 1 displaying macrocephaly, broad forehead, deep set eyes, broad nasal root, hanging columella and short philtrum. b Pedigree for patient 1. c. Pedigree for patient 2
On examination the patient was macrocephalic with OFC of 59 cm (>97th centile). His facial features appeared similar to his mother with broad forehead, deep set eyes, broad nasal root, prominent columella, and short philtrum (Fig. 2a). Poor eye contact, low attention span and motor stereotypies were observed. There were no neurocutaneous markers and his neurological examination was grossly normal. Standardized psychological assessment could not be completed due to limited attention for directed tasks and his gross cognitive testing indicated his functioning at 25-month-old level. His past evaluation included normal MRI of Brain, EEG and negative fragile X testing.
Patient 2 is a now 3-year-old boy who presented for genetic evaluation due to global developmental delay and congenital anomalies. His parents were non-consanguineous and of mixed ethnicity (Fig. 2c). Both parents are generally healthy and had unremarkable developmental histories. His birth weight 2.81 kg (13th percentile), length 49 cm (32nd percentile) and OFC of 32.1 cm (3rd percentile). Cutis aplasia was found after delivery. He was noted to have a large, irregular skin defect on his posterior scalp with some islands of normal skin. The calvarium in the affected region was thin. Three stellate shaped ulcerations involving vertex and occiput of the scalp were noted (largest measuring approximately 4 cm × 3 cm; the next smallest measuring approximately 3.5 cm × 2.5 cm; and the smallest approximately 1 cm in diameter). The scalp defect was managed conservatively and healed with scarring and patchy alopecia. At 3 years of age he had predominant language delay and lagging behind in his head circumference for age. Examination revealed OFC of 46 cm (<3rd percentile). Scalp was notable for extensive scarring and patchy alopecia. Ears were cupped and mildly simplified. He had broad forehead, hypertelorism with arched eyebrows and a slightly upturned nasal tip. Neurological examination was unremarkable except for expressive language delay. Previous genetic testing included a normal chromosome microarray analysis.
Genetic analysis
Patient 1 had exome sequencing performed through a CLIA-certified laboratory. A c.77C > A, p.(Ser26*) pathogenic nonsense variant was reported in TRIO (NM_007118.2). This variant was inherited from the patient’s affected mother. This variant has not been previously reported in gnomAD, nor has it been previously associated with a disease phenotype based on ClinVar or published literature. It is located in exon 1 and would cause truncation in the n-terminal SEC14 domain which lies upstream of the spectrin repeats. In silico prediction of NMD suggests this variant would cause the affected transcript to be targeted for degradation [40]. The function of the SEC14 domain has not been well established other than a role in subcellular targeting and possibly the regulation of endosome dynamics [41]. Patient 1 also had a c.11G > C, p.(Arg4Pro) missense variant of uncertain significance (VUS) reported in PIK3CA (NM_006218.2). This variant was also maternally inherited. Due to lack of phenotypic overlap with the associated MCAP syndrome, CLOVE syndrome, and fibroadipose hypoplasia, this variant was not thought to be contributing to the patient’s phenotype.
Patient 2 had exome sequencing performed through the same clinical laboratory as patient 1. A c.2690dupA, p.(Gln898Alafs*61) pathogenic frameshift variant was reported in TRIO (NM_007118.2). The variant was not inherited from the unaffected mother and a paternal sample was not available. This variant has not been previously reported in gnomAD, nor has it been previously reported in ClinVar or published literature. It is located in exon 15 and would cause an early truncation in the spectrin repeats. In silico prediction of NMD also suggest this variant would cause any affected transcript to be targeted for degradation [40].
Discussion and conclusions
Here we report on two patients with novel pathogenic presumed LOF variants reported in TRIO. Both patients have phenotypes overlapping with previously reported patients including severe (patient 1) and moderate (patient 2) developmental delay, and ID. Both had dysmorphisms including broad/high forehead, deep set eyes, broad nasal root, prominent columella and short philtrum in patient 1 and cupped ears, broad forehead, hypertelorism, arched eyebrows and a slightly upturned nasal tip in patient 2. Of note, Ba et al. also reported several patients with high forehead in their cohort [22]. A novel phenotype presenting in patient 2 includes extensive cutis aplasia which has not been previously reported and may represent an expanded phenotype associated with this disorder.
Microcephaly was reported in patient 2 which matches well with most previously characterized patients with nonsense/truncating variants (including p.(Arg217*), p.(Gln768*), p.(Asp1251Valfs*11), p.(Trp1376*), p.(Gln1489Argfs*11), p.(Arg1620Serfs*10), p.(Val1698Leufs*61), p.(Lys1918*), p.(Leu2031Phefs*9), and p.(Phe2472Serfs*54)) [16, 22, 35,36,37]. Notably, patient 2 does not show a severe ID phenotype or macrocephaly as is reported for patients with missense variants in the same spectrin repeat domain, reinforcing that microcephaly is associated with the majority of presumed LOF variants and thereby with haploinsufficiency of the gene.
Interestingly, patient 1 had macrocephaly in contrast to the majority of other patients with truncating and presumed LOF variants. Barbosa et al. described a group of non-related individuals with macrocephaly who had missense variants clustering on the spectrin repeat domain. The authors performed in vitro experiments and showed that the missense variants at the spectrin repeat 7 cause a gain-of-function effect, inducing hyperactivation of RAC1. None of the individuals with presumable LOF variants in their cohort presented with macrocephaly [36]. One possibility as to why patient 1 seems to have a different phenotype may be that the variant is in the far N-terminal region and therefore there are several transcripts with downstream alternate start sites that could be unaffected by this variant (ENST00000509967.2, ENST00000513206.1, ENST00000344135.5). Variations in the location and timing of expression of these alternate transcripts may explain the discrepancy in observed phenotypes between patient 1 and other patients with presumed LOF variants.
Overall, we demonstrate two cases with patients presenting with neurological phenotypes and reported pathogenic, presumed LOF variants in TRIO. We add to the known clinical phenotype spectrum associated with this gene with the report of cutis aplasia in patient 2. We also add to the literature the third documented case of familial autosomal dominant inheritance of a TRIO-associated disorder highlighting the variable expressivity associated with TRIO disorders. Moreover, we report on a patient with macrocephaly and a nonsense variant upstream to the domain hotspot that has been associated with this phenotype. Future work clarifying the status of transcript degradation/protein translation for truncating and nonsense variants in patient samples, especially testing for hyperactivation of RAC1 in the individual with a nonsense variant is needed to fully characterize the effect of these types of variants.
Availability of data and materials
Genomic sequence data cannot be deposited due to restrictions imposed by the IRB. The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to their containing information that could compromise the privacy of research participants.
Abbreviations
- TRIO:
-
Triple functional domain
- GEF:
-
Guanine nucleotide exchange factor
- ID:
-
Intellectual disability
- ASD:
-
Autism spectrum disorder
- OFC:
-
Occipitofrontal circumference
- NMD:
-
Nonsense-mediated decay
- LOF:
-
Loss-of-function
- AD:
-
Autosomal dominant
- HGVS:
-
Human Genome Variation Society
References
Schmidt S, Debant A. Function and regulation of the rho guanine nucleotide exchange factor trio. Small GTPases. 2014;5:e29769.
Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–35.
Debant A, Serra-Pages C, Seipel K, O'Brien S, Tang M, Park SH, Streuli M. The multidomain protein trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci U S A. 1996;93(11):5466–71.
Govek EE, Newey SE, Van Aelst L. The role of the rho GTPases in neuronal development. Genes Dev. 2005;19(1):1–49.
Cannet A, Schmidt S, Delaval B, Debant A. Identification of a mitotic Rac-GEF, trio, that counteracts MgcRacGAP function during cytokinesis. Mol Biol Cell. 2014;25(25):4063–71.
Ba W, van der Raadt J, Nadif Kasri N. Rho GTPase signaling at the synapse: implications for intellectual disability. Exp Cell Res. 2013;319(15):2368–74.
Herring BE, Nicoll RA. Kalirin and trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc Natl Acad Sci U S A. 2016;113(8):2264–9.
Bellanger JM, Lazaro JB, Diriong S, Fernandez A, Lamb N, Debant A. The two guanine nucleotide exchange factor domains of trio link the Rac1 and the RhoA pathways in vivo. Oncogene. 1998;16(2):147–52.
Blangy A, Vignal E, Schmidt S, Debant A, Gauthier-Rouviere C, Fort P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J Cell Sci. 2000;113(Pt 4):729–39.
Chhatriwala MK, Betts L, Worthylake DK, Sondek J. The DH and PH domains of trio coordinately engage rho GTPases for their efficient activation. J Mol Biol. 2007;368(5):1307–20.
Jaiswal M, Dvorsky R, Ahmadian MR. Deciphering the molecular and functional basis of Dbl family proteins: a novel systematic approach toward classification of selective activation of the rho family proteins. J Biol Chem. 2013;288(6):4486–500.
McPherson CE, Eipper BA, Mains RE. Multiple novel isoforms of trio are expressed in the developing rat brain. Gene. 2005;347(1):125–35.
Portales-Casamar E, Briancon-Marjollet A, Fromont S, Triboulet R, Debant A. Identification of novel neuronal isoforms of the rho-GEF trio. Biol Cell. 2006;98(3):183–93.
Yoshizuka N, Moriuchi R, Mori T, Yamada K, Hasegawa S, Maeda T, Shimada T, Yamada Y, Kamihira S, Tomonaga M, et al. An alternative transcript derived from the trio locus encodes a guanosine nucleotide exchange factor with mouse cell-transforming potential. J Biol Chem. 2004;279(42):43998–4004.
Ma XM, Huang JP, Eipper BA, Mains RE. Expression of trio, a member of the Dbl family of rho GEFs in the developing rat brain. J Comp Neurol. 2005;482(4):333–48.
Katrancha SM, Wu Y, Zhu M, Eipper BA, Koleske AJ, Mains RE. Neurodevelopmental disease-associated de novo mutations and rare sequence variants affect TRIO GDP/GTP exchange factor activity. Hum Mol Genet. 2017;26(23):4728–40.
Peng YJ, He WQ, Tang J, Tao T, Chen C, Gao YQ, Zhang WC, He XY, Dai YY, Zhu NC, et al. Trio is a key guanine nucleotide exchange factor coordinating regulation of the migration and morphogenesis of granule cells in the developing cerebellum. J Biol Chem. 2010;285(32):24834–44.
Zong W, Liu S, Wang X, Zhang J, Zhang T, Liu Z, Wang D, Zhang A, Zhu M, Gao J. Trio gene is required for mouse learning ability. Brain Res. 1608;2015:82–90.
O'Brien SP, Seipel K, Medley QG, Bronson R, Segal R, Streuli M. Skeletal muscle deformity and neuronal disorder in trio exchange factor-deficient mouse embryos. Proc Natl Acad Sci U S A. 2000;97(22):12074–8.
Katrancha SM, Shaw JE, Zhao AY, Myers SA, Cocco AR, Jeng AT, Zhu M, Pittenger C, Greer CA, Carr SA, et al. Trio Haploinsufficiency causes neurodevelopmental disease-associated deficits. Cell Rep. 2019;26(10):2805–17 e2809.
Tao T, Sun J, Peng Y, Wang P, Chen X, Zhao W, Li Y, Wei L, Wang W, Zheng Y, et al. Distinct functions of trio GEF domains in axon outgrowth of cerebellar granule neurons. J Genet Genomics. 2019;46(2):87–96.
Ba W, Yan Y, Reijnders MR, Schuurs-Hoeijmakers JH, Feenstra I, Bongers EM, Bosch DG, De Leeuw N, Pfundt R, Gilissen C, et al. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum Mol Genet. 2016;25(5):892–902.
Carraro M, Monzon AM, Chiricosta L, Reggiani F, Aspromonte MC, Bellini M, Pagel K, Jiang Y, Radivojac P, Kundu K, et al. Assessment of patient clinical descriptions and pathogenic variants from gene panel sequences in the CAGI-5 intellectual disability challenge. Hum Mutat. 2019;40(9):1330–45.
Geisheker MR, Heymann G, Wang T, Coe BP, Turner TN, Stessman HAF, Hoekzema K, Kvarnung M, Shaw M, Friend K, et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat Neurosci. 2017;20(8):1043–51.
Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, Karczewski KJ, Cutler DJ, Devlin B, Roeder K, et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet. 2017;49(4):504–10.
Deciphering Developmental Disorders S. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542(7642):433–8.
Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–21.
Jansen S, Hoischen A, Coe BP, Carvill GL, Van Esch H, Bosch DGM, Andersen UA, Baker C, Bauters M, Bernier RA, et al. A genotype-first approach identifies an intellectual disability-overweight syndrome caused by PHIP haploinsufficiency. Eur J Hum Genet. 2018;26(1):54–63.
Eising E, Carrion-Castillo A, Vino A, Strand EA, Jakielski KJ, Scerri TS, Hildebrand MS, Webster R, Ma A, Mazoyer B, et al. A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol Psychiatry. 2019;24(7):1065–78.
Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, Turner TN, Liu Y, Zhao W, Hoekzema K, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. 2016;7:13316.
de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–9.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–41.
Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, Thiruvahindrapuram B, Merico D, Jobling R, Nalpathamkalam T, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20(4):435–43.
Boonsawat P, Joset P, Steindl K, Oneda B, Gogoll L, Azzarello-Burri S, Sheth F, Datar C, Verma IC, Puri RD, et al. Elucidation of the phenotypic spectrum and genetic landscape in primary and secondary microcephaly. Genet Med. 2019;21(9):2043–58.
Kahrizi K, Hu H, Hosseini M, Kalscheuer VM, Fattahi Z, Beheshtian M, Suckow V, Mohseni M, Lipkowitz B, Mehvari S, et al. Effect of inbreeding on intellectual disability revisited by trio sequencing. Clin Genet. 2019;95(1):151–9.
Barbosa S, Greville-Heygate S, Bonnet M, Godwin A, Fagotto-Kaufmann C, Kajava AV, Laouteouet D, Mawby R, Wai HA, Dingemans AJM, et al. Opposite modulation of RAC1 by mutations in TRIO is associated with distinct, domain-specific neurodevelopmental disorders. Am J Hum Genet. 2020;106(3):338–55.
Pengelly RJ, Greville-Heygate S, Schmidt S, Seaby EG, Jabalameli MR, Mehta SG, Parker MJ, Goudie D, Fagotto-Kaufmann C, Mercer C, et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J Med Genet. 2016;53(11):735–42.
Sadybekov A, Tian C, Arnesano C, Katritch V, Herring BE. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of trio. Nat Commun. 2017;8(1):601.
Chen SY, Huang PH, Cheng HJ. Disrupted-in-schizophrenia 1-mediated axon guidance involves TRIO-RAC-PAK small GTPase pathway signaling. Proc Natl Acad Sci U S A. 2011;108(14):5861–6.
Coban-Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, Bayram Y, Chinn IK, Karaca E, Punetha J, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. 2018;103(2):171–87.
Saito K, Tautz L, Mustelin T. The lipid-binding SEC14 domain. Biochim Biophys Acta. 2007;1771(6):719–26.
Acknowledgements
We would like to thank the families for participating in this research.
Consent to participate
Parents of all of the participants provided written consent to participate in the study.
Funding
This work was supported by the Mayo Clinic Center for Individualized Medicine (CIM), the Investigative and Functional Genomics Program and the William O. Lund, Jr. and Natalie C. Lund Charitable Foundation.
Author information
Authors and Affiliations
Contributions
All authors read and approved the manuscript. LSR performed variant analysis and wrote the manuscript. KM performed clinical examination and care of the patients, contributed to writing, intellectual content and revision of the manuscript. FPV assisted in variant analysis and editing of the manuscript. EWK supported this research and contributed to writing and editing of the manuscript. BL provided clinical examination and care of the patients, contributed to writing, intellectual content and critical revision of the manuscript. All the authors approved the submitted version and the modified version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Mayo Clinic institutional review board. IRB#: 12–009346. Written consent was obtained from the parents or legal guardians of all participants since the patients were under the age of 16.
Consent for publication
Written informed consent for publication of identifying images and other personal or clinical details was obtained from the parents or legal guardians since the participants were under the age of 18. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Schultz-Rogers, L., Muthusamy, K., Pinto e Vairo, F. et al. Novel loss-of-function variants in TRIO are associated with neurodevelopmental disorder: case report. BMC Med Genet 21, 219 (2020). https://doi.org/10.1186/s12881-020-01159-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01159-y