
Abstract
Background
Okur-Chung neurodevelopmental syndrome (OCNDS) and tricho-rhino-phalangeal syndrome type I (TRPSI) are rare Mendelian diseases. OCNDS is caused by CSNK2A1 gene variants and TRPSI is caused by the TRPS1gene. However, to have two Mendelian diseases in one patient is even rarer.
Case presentation
A 6-year-10-month-old boy characterized by special facial features, short stature and mental retardation was referred to our pediatric endocrinology department. Whole-exome sequencing (WES) was done to detect the molecular basis of his disease. This patient was confirmed to carry two variants in the CSNK2A1 gene and one in the TRPS1 gene. The variant in the CSNK2A1 gene was vertically transmitted from his father, and the variant in TRPS1 gene from his mother. These two variants are classified as pathogenic and the causes of the presentation in this child. This patient’s father and mother have subsequently been diagnosed as having OCNDS and TRPSI respectively.
Conclusion
This is the first reported case of a dual molecular diagnosis of tricho-rhino-phalangeal syndrome type I and Okur-Chung neurodevelopmental syndrome in the same patient. This patient is the first published example of vertical transmission of this recurrent CSN2A1 variant from parent to child. A novel variant in the TRPS1 gene that is pathogenic was also identified. In conclusion, identification of the variants in this patient expands the phenotypes and molecular basis of dual Mendelian diseases.
Similar content being viewed by others


Background
The tricho-rhino-phalangeal syndrome (TRPS) was first described by Andres Giedion in 1966 [1]. It is a rare autosomal dominant disorder characterized by distinctive facial features and skeletal abnormalities, such as sparse scalp hair, a peculiar shaped nose, cone-shaped epiphyses of the phalanges, and short stature [2]. TRPS comprises three subtypes, TRPS I (OMIM #190350), TRPS II (OMIM #150230) and TRPS III (OMIM #190351) depending on clinical presentation and molecular differences. TRPS I and TRPS III are caused by heterozygous mutations of the TRPS1gene. TRPS III differs from TRPSI by the presence of severe brachydactyly due to short metacarpals and severe short stature [3]. TRPS II is caused by a contiguous gene deletion of TRPS1, RAD21, and EXT1 with multiple osteochondromas, which is absent in TRPS I patients [4, 5]. TRPS1 gene encode a zinc-finger, GATA-type transcription factor, which is involved in the development and differentiation of the bones, kidneys, and hair follicles [2].
Okur–Chung neurodevelopmental syndrome (OCNDS, OMIM #617062) is an autosomal-dominant disorder which is characterized by intellectual disability, developmental delay, behavioral problems, and other multisystemic abnormalities. It is caused by CSNK2A1 gene variants which was first reported by Okur in 2016 [6]. CSNK2A1 gene is expressed in the brain and encodes the catalytic subunit of protein kinase CK2, which is in involved in many biologic processes.
Usually, it is hard to diagnose these rare Mendelian diseases through routine clinical practice such as the recognition of phenotypes, biochemical tests and imaging. Whole-exome sequencing (WES) provides a way to reveal the relationship between clinical phenotypes and genotypes. Recent studies show that 4.9% (101/2076) of patients have had more than one molecular diagnosis [7]. In this case study, a 6-year-10-month-old boy characterized with special facial features, short stature and mental retardation was confirmed to be carrying compound variants in both the CSNK2A1 gene and also the TRPS1 gene using WES. This patient has a dual molecular diagnosis of tricho-rhino-phalangeal syndrome type I and Okur-Chung neurodevelopmental syndrome.
Case presentation
Patient and clinical evaluation
A 6-year-10-month-old boy was referred to the pediatric endocrinology department with short stature and developmental delay. This boy was the first child of non-consanguineous parents. He was born at full term. His birth length was 48 cm (− 1.33 s.d.) and birth weight was 2.65 kg (− 1.80 s.d.). His mother noted he had poor growth after the age of 1, but didn’t know the exact growth velocity. His developmental milestones were delayed. He first walked by himself and spoke at 4 years of age. He also suffered from constipation. At 6 years, he started primary school, but struggled to socially adapt to his peer group. He was unable to express phrases and communicate with other children of his age. His father is 147 cm (− 4.01 s.d.) tall, with unusual facial features such as a round face, broad nasal bridge, short upturned nose and arched eyebrows. He has mild intellectual disability. Paternal grandparents’ heights are 160 cm and 150 cm, both are of normal intelligence. The patient’s mother is 141 cm (− 3.63 s.d.) tall, with facial features such as sparse scalp hair and a pear-shaped nose. She is of normal intelligence. However, maternal grandparents’ details are unknown due to adoption.
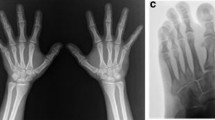
At the time of presentation at 6 years 10 months, his height was 94.5 cm (− 5.8 s.d.), his weight was 12.8 kg (− 4.3 s.d.), and his head circumference was 50.1 cm (− 1.0 s.d.). Physical examination showed facial dysmorphic features such as sparse scalp hair, a pear-shaped nose, long flat philtrum, thin upper vermillion border and short stature (Fig. 1). Standard blood and urine screening tests were normal, and a growth hormone (GH) provocation test with arginine/levodopa showed appropriate GH peaks. No disturbances of calcium/phosphate metabolism were found. The bone age, performed according to the atlas of Greulich and Pyle was significantly delayed (bone age 2 years at 6 years and 10 months chronological age). Echocardiography was normal. MRI of the brain and pituitary region revealed a reduced size anterior pituitary gland. On the Wechsler Intelligence Scale for Children (Chinese Revision) (WISC-RC) he was found to perform at the level of severe intellectual disability (Intelligent Quotient < 40).

Distinctive face and hands of the patient in this study. Facial dysmorphic features include sparse scalp hair, a pear-shaped nose, long flat philtrum and thin upper vermillion border
His parents have given informed consent for his pictures and medical data to be published.
WES analysis and sanger sequencing
Peripheral blood samples were collected from the patient and his parents after obtaining written informed consent. Single WES was done on this patient’s blood, then Sanger sequencing was performed on his parents’ blood samples to verify the variants. The genomic DNA was extracted from peripheral blood samples by using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. 1.5 μg genomic DNA was fragmented into an average size of 300 bp, then the fragmented genomic DNA was used for preparation of sequencing libraries. 8 bp barcoded sequencing adaptors were then ligated with DNA fragments before final hybridization with xGen® Exome Research Panel v1.0 focused exon probes (IDT, USA). All the exons were sequenced by Illumina HiSeq X-10 platform. Purified sequencing libraries were pooled together and massively parallel sequenced by Illumina HiSeq X platform to produce a sequencing yield of 10.0 Gb raw data. The reads were mapped to the reference human genome using NextGene software (SoftGenetics LLC, State College, PA). All the variants were compared with databases such as 1000 Genomes Project, the Exome Aggregation Consortium (ExAC) and dbSNP. The variants with a minor allele frequency greater than 0.01 in the control databases were excluded. Pathogenic predication analysis was carried out on platforms of SIFT, Polyphen-2 and mutation taster. All the variants were studied to confirm if they were located in the conserved gene region and if the variants affected the protein function or structure. Following these filtering steps, potential variants were considered to cause disease and were subsequently validated by Sanger sequencing. The mean read depth of this patient against RefSeq protein-coding regions was 93.24 reads with 97.71% being covered by ⩾20 reads, and 93.70% covered by ⩾30 reads.
We identified a CSNK2A1 variant (NM_177559: c.593 A > G, p.K198R) and a TRPS1 variant (NM_014112:c.2174delA, p.N725fs), which were vertically transmitted from his father and mother, respectively (Fig. 2). The variant c.593 A > G(p.K198R, rs 869,312,840) on CSNK2A1 gene has previously been reported in the medical literature as ‘pathogenic’ on at least six previous occasions (PS4-Strong) [8,9,10,11]. This variant is in the critical protein domain (PM1) and it is extremely rare in the general population (PM2). The patient’s phenotype is consistent and specific for Okur–Chung neurodevelopmental syndrome (PP4). According to American College of Medical Genetics and Genomics/The Association for Molecular Pathology (ACMG/AMP) variant interpretation guidelines [12], this variant can be classified as pathogenic. The variant c.2174delA(p.N725fs) on TRPS1 was a novel variant, which causes a frame shift. This variant affects the amino acid residue number 725 and introduces a premature stop codon after 39 residues in the gene sequence. This is a loss of function variant (PVS1), which is absent in general population (PM2). The patient’s and his mother’s phenotypes are highly specific for TRPS I (PP4). This can be classified as pathogenic based on ACMG/AMP guidelines [12].

a Patient’s family’s pedigree. Black arrow shows the proband. Black square indicates the patient affected with OCNDS caused by a variant in the CSNK2A1 gene. Square filled with transverse lines indicates the patient affected with TRPS I caused by a variant in the TRPS1 gene. The compound heterozygote found in this patient was vertically transmitted from his father and mother respectively; b Validation by Sanger sequencing of CSNK2A1 and TRPS1 gene in this family
Discussion and conclusions
In this report we describe a dual molecular diagnosis of OCNDS and TRPS1 in a Chinese patient. This patient has inherited both clinical presentations of OCNDS and TRPS I (Table 1), and his father and mother present typical manifestations of OCNDS and TRPS I respectively.
OCNDS is a new syndrome, which was first described in 2016 by Okur et al. [6]. Common features included development delay, intellectual disability, variable dysmorphic facial features, gastrointestinal problems and musculoskeletal abnormalities. Patients may present with microcephaly, low-set and folded ears, arched eyebrows, epicanthal folds, broad nasal bridge, upturned nose, or a thin upper lip. To date, no more than 60 individuals with variants in CSNK2A1 have been reported [8,9,10,11, 13, 14]. Most of these variants are de novo variants. The variant detected in this patient was vertically transmitted from his father, who also presents with clinical features of OCNDS. Actually, this is the first OCNDS family report. The variant c.593 A > G (p.K198R) is common in OCNDS, which was previously described in the literature in several other affected patients, so it is thought to be a “hot spot” [8,9,10,11]. Interestingly, this patient is the first published example of vertical transmission of this recurrent CSN2A1 variant from parent to child. There is some overlap between these clinical reports and our patient, such as delayed development and intellectual disability, but not every affected individual has growth retardation as in our patient, some may actually be of normal height (above − 2.0 s.d.).
TRPS I was first described in 1966, and can be diagnosed based on clinical criteria [1]. This patient fulfill sufficient features to meet the clinical criteria for this diagnosis with the presence of sparse scalp hairs, a bulbous and pear shaped nose, long philtrum and brachydactyly. His mother had similar features. Maas SM et al. [15] described the phenotype and genotype in 103 patients with TRPS. Decreased linear growth was observed in almost all patients. It is more obviously postnatally than prenatally, and half of the adult patients are below − 2 s.d. in height. Growth hormone supplementation has been used in patients with TRPS; however the response to GH therapy is variable in affected individuals [16]. The percentage of TRPS I individuals with intellectual disability is similar to that in the general population [15], so the reason that the described patient has low IQ score could be explained by his also affected by OCNDS. Almost all patients have cone-shaped epiphyses, which can also be detected at an early age. However, our patient doesn’t show this characteristic radiological feature, consistent with the variable clinical presentation of TRPS.
Molecular methods confirmed the diagnosis of TRPS I in this patient and his mother. The TRPS1 gene contains 7 exons, and encodes 1294 amino acids. Most of the mutations are nonsense and frameshift mutations which are located in exons 4–7 of the gene. The number of recurrent mutations is very low [15]. Variant c.2174delA(p.N725fs) is novel, located in exon 5. This alters the TRPS1 reading frame and results in the introduction of a premature stop codon.
Co-occurrence of two Mendelian diseases in one patient is very rare. Posey et al. [7] summarized a retrospective analysis of data from 7374 patients who had been referred for whole exome sequencing. Amongst these patients, 2076 (28.2%) had at least one molecular diagnosis, but only 101 (4.9%) were diagnosed with two or more disease loci. In the same patient, the two Mendelian diseases could be distinct or overlapping by phenotype.
Our patient has inherited two variants of the CSNK2A1 gene and the TRPS1 gene separately from his father and mother. These two syndromes share the same presentation “short stature”. However, compared with other patients with single OCNDS or TRPS I, and with his father (− 5.8 s.d. VS − 4.01 s.d.) or his mother (− 5.8 s.d. VS − 3.63 s.d.), this patient who has dual diagnosis of OCNDS and TRPS I, has a more significant degree of short stature. These two pathogenic variants of the CSNK2A1 and TRPS1 genes perhaps have a combined exaggerated deleterious effect on linear growth. Whilst these two syndromes have different facial features, this patient resembles his mother.
We referral the patient for speech and language therapy. We suggested growth hormone therapy to improve short stature, but the family rejected this option for economic reasons.
In summary, our study describes a Chinese patient who suffered from simultaneous tricho-rhino-phalangeal syndrome type I and Okur-Chung neurodevelopmental syndrome. The variants in CSNK2A1 gene were vertically transmitted from his father and a variant in TRPS1 gene from his mother. The recurrent variant c.593 A > G(p.K198R) in the CSNK2A1 gene has not previously been described in two members of the same family. A novel variant c.2174delA(p.N725fs) in the TRPS1 gene that is pathogenic was also identified. Further functional analysis of this variant could expand the mechanism exploration of TRPS I. Dual Mendelian diseases in one patient is rare; however, with the availability of molecular diagnostic methodology, we may be able to identify more patients with multiple molecular diagnoses, and in turn, improve knowledge about inherited human diseases.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on request.
Abbreviations
- WES:
-
Whole-exome sequencing
- TRPS:
-
Tricho-rhino-phalangeal syndrome
- OCNDS:
-
Okur–Chung neurodevelopmental syndrome
- GH:
-
Growth hormone
- s.d.:
-
Standard Deviation
References
Giedion A. Das Tricho-rhino-phalangeal Syndrom. Helv Paediat Acta. 1966;21:475–82.
Momeni P, Glockner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, Hennekam R, Meinecke P, Zabel B, Rosenthal A, Horsthemke B, Ludecke H-J. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet. 2000;24:71–4.
Lüdecke HJ, Schaper J, Meinecke P, Momeni P, Gross S, von Holtum D, et al. Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet. 2001;68(1):81–91.
Hilton MJ, Sawyer JM, Gutierrez L, et al. Analysis of novel and recurrent mutations responsible for the tricho–rhino–phalangeal syndromes. J Hum Genet. 2002;47:103–6.
Hou J, Parrish J, Ludecke H-J, Sapru M, Wang Y, Chen W, Hill A, Siegel-Bartelt J, Northrup H, Elde FFB, Chinault C, Horsthemke B, Wagner MJ, Wells DE. A 4-megabase YAC contig that spans the Langer-Giedion syndrome region on human chromosome 8q24.1: use in refining the location of the trichorhinophalangeal syndrome and multiple exostoses genes (TRPS1 and EXT1). Genomics. 1995;29:87–97.
Okur V, Cho MT, Henderson L, Retterer K, Schneider M, Sattler S, et al. De novo mutations in CSNK2A1 are associated with neurodevelopmental abnormalities and dysmorphic features. Hum Genet. 2016;135:699–705.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376(1):21–31.
Nakashima N, Tohyama J, Nakagawa E, Watanabe Y, Siew CG, Kwong CS, et al. Identification of de novo CSNK2A1 and CSNK2B variants in cases of global developmental delay with seizures. J Hum Genet. 2019;64(4):313–22.
Owen C, Bowden R, Parker MJ, Patterson J, Patterson JO, Price S, et al. Extending the phenotype associated with the CSNK2A1-related Okur–Chung syndrome—a clinical study of 11 individuals. Am J Med Gene A. 2018;176(5):1108–14.
Chiu ATG, Pei SLC, Mak CCY, Leung GKC, Yu MHC, Lee SL, et al. Okur-Chung neurodevelopmental syndrome: eight additional cases with implications on phenotype and genotype expansion. Clin Genet. 2018;93(4):880–90.
Trinh J, Huning I, Budler N, Hingst V, Lohmann K, Gillessen G. A novel de novo mutation in CSNK2A1: reinforcing the link to neurodevelopmental abnormalities and dysmorphic features. J Hum Genet. 2017;62(11):1005–6.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, ACMG Laboratory Quality Assurance Committee, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Duan HL, et al. A case of Okur-Chung syndrome caused by CSNK2A1 gene variation and review of literature. Zhonghua Er Ke Za Zhi. 2019;57(5):368–72.
Akahira-Azuma M, Tsurusaki Y, Enomoto Y, Mitsui J, Kurosawa K. Refifining the clinical phenotype of Okur–Chung neurodevelopmental syndrome. Hum Genome Var. 2018;5:18011.
Maas SM, Shaw AC, Bikker H, Ludecke HJ, Tuin K, Badura-Stronka M, et al. Phenotype and genotype in 103 patients with tricho-rhino-phalangeal syndrome. Eur J Med Genet. 2015;58(5):279–92.
Merjaneh L, Parks JS, Muir AB, Fadoju D. A novel TRPS1 gene mutation causing trichorhinophalangeal syndrome with growth hormone responsive short stature: a case report and review of the literature. Int J Pediatr Endocrinol. 2014;2014(1):16.
Acknowledgments
We acknowledge all the patients and families for permission to publish this case.
Funding
No external funding was necessary for this study.
Author information
Authors and Affiliations
Contributions
QL and JZW participated in the design and coordination of the study. SSX performed the laboratory analyses and drafted the manuscript. LLL and JS performed the statistical analysis. All authors provided input into the final manuscript. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethical Committee of The First Affiliated Hospital of Xiamen University approved the study.
Consent for publication
A written informed consent for publication of clinical details and photographs was obtained from this patient’s parents.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xu, S., Lian, Q., Wu, J. et al. Dual molecular diagnosis of tricho-rhino-phalangeal syndrome type I and Okur-Chung neurodevelopmental syndrome in one Chinese patient: a case report. BMC Med Genet 21, 158 (2020). https://doi.org/10.1186/s12881-020-01096-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01096-w